 去看看
去看看

一、癫痫发作
癫痫发作(epileptic seizure)是指脑神经元异常过度、同步化放电活动所造成的短暂、一过性临床表现。
癫痫发作具有三方面要素:
1.临床表现
癫痫发作必须有临床表现(症状和/或体征)。临床表现可多种多样,如感觉﹑运动﹑自主神经﹑知觉﹑情感﹑认知及行为等障碍。
2.起始和终止的形式
癫痫发作一般具有突发突止﹑短暂一过性、自限性的共同特点。通常可以根据行为表现或脑电图改变来判断癫痫发作的起始和终止。癫痫持续状态是一种表现为持续或反复发作的特殊情况。
3.脑部异常
过度同步化放电要通过脑电图检查才能证实。这是癫痫发作区别于其他发作性症状的最本质的特征。
按照有无急性诱因,癫痫发作大体上可分为诱发性发作(provoked seizure)和非诱发性发作(unprovoked seizure)。诱发性发作最常见于中枢神经系统疾病(感染/卒中等)或全身系统性疾病(血糖异常/电解质紊乱/中毒/发热等)的急性期,是一种急性症状性发作(acute symptomatic seizure)。这种发作仅代表疾病急性期的一种症状,不意味急性期过后一定反复出现癫痫发作。非诱发性发作则没有明确的急性诱因。例如,病毒性脑炎急性期出现的癫痫发作是诱发性发作,而脑炎数年后出现的癫痫发作则为非诱发性发作。
二、癫痫
癫痫(epilepsy)是一种以具有持久性的致痫倾向为特征的脑部疾病。癫痫不是单一的疾病实体,而是一种有着不同病因基础、临床表现各异但以反复癫痫发作为共同特征的慢性脑部疾病状态。
三、癫痫综合征
癫痫综合征(epilepticsyndrome)指由一组特定的临床表现和脑电图改变组成的癫痫疾患(即脑电-临床综合征)。
临床上常结合发病年龄、发作类型、病因学、解剖基础、发作时间规律、诱发因素、发作严重程度、其他伴随症状、脑电图及影像学结果、既往史、家族史、对药物的反应及转归等资料,作出某种癫痫综合征的诊断。诊断癫痫综合征对于治疗选择、判断预后等方面具有一定指导意义。
四、癫痫相关脑病
癫痫患者除了癫痫性异常,还可以出现不同程度的以神经精神功能障碍或退化为特征的脑病表现,包括认知、语言、感觉、运动及行为等方面。脑病表现可为全面性或具有选择性。根据患者脑病与癫痫的关系,可以分为3类:第1类,由于癫痫性异常本身(即频繁癫痫发作和/或癫痫样放电)造成的脑病,称为癫痫性脑病(epileptic encephalopathy);第2类,如果癫痫患者伴有由于潜在发育性异常病因所致的脑病,癫痫发作本身对于脑病没有或者不起主要的作用,则称为癫痫伴发育性脑病(developmental encephalopathy);第3类,癫痫患者的脑病状态是由潜在发育性异常病因和癫痫性异常双重作用导致的,此时称为发育性癫痫性脑病(developmental and epileptic encephalopathy),本组疾患大多为新生儿、婴幼儿或儿童期发病,脑电图明显异常,药物治疗效果差,临床常见韦斯特综合征(West syndrome)、伦诺克斯-加斯托综合征(Lennox-Gastaut syndrome,LGS)及德拉韦综合征(Dravet syndrome)等均属于此类疾患。
一、癫痫诊断的原则及流程
癫痫诊断的原则和完整流程可分为五个步骤。
(一)确定发作性事件是否为癫痫发作
涉及发作性事件的鉴别,包括诱发性癫痫发作和非诱发性癫痫发作的鉴别。
(二)确定癫痫发作的类型
按照ILAE癫痫发作分类来确定。
(三)确定癫痫及癫痫综合征的类型
按照ILAE癫痫及癫痫综合征分类系统来确定。有些病例无法归类于某种特定癫痫综合征。
(四)确定病因
(五)确定残障(disability)和共患病(co-morbidity)
二、癫痫诊断的标准
传统上,临床出现两次(间隔至少24小时)非诱发性癫痫发作时就可诊断癫痫。这是目前普遍采用的、具有临床可操作性的诊断标准。
2014年ILAE癫痫临床实用性定义指出,除了上述传统的诊断标准,对于如下两种情况也可考虑诊断癫痫:
1.首次非诱发性(或反射性)发作,并且在未来10年内再次发作风险至少达到60%。
这种情况对于首次发作就尽早诊断并控制癫痫具有积极意义,但多数情况下较难确定某个体首次发作后的具体再发风险。目前有限证据提示,能够增加成人首次癫痫发作后再发风险的因素包括:①存在既往脑损伤病史;②脑电图有痫样异常表现;③脑部影像学存在致痫病变;④首次发作为夜间发作。
2.诊断某种癫痫综合征 鉴于及时摘掉癫痫诊断“标签”意义重大,2014年ILAE癫痫临床实用性定义同时也指出了可解除癫痫诊断(epilepsy resolved)的两种情况:
(1)已经超过了某种年龄依赖癫痫综合征的患病年龄。
(2)已经10年无发作,并且近5年已停用抗癫痫发作药。
三、癫痫诊断的方法
临床上,完整的癫痫诊断(五步骤流程)通常需要获得如下信息:
(一)病史资料
完整病史是癫痫诊断中最重要的环节。应包括:现病史(重点是发作史)、出生史、既往史、家族史、疾病的社会心理影响等(表2-1)。
表2-1 癫痫诊断中的重要病史资料

| 现病史 首次发作年龄 发作前状态或促发因素(觉醒、清醒、睡眠、饮酒、少眠、过度疲劳、心理压力、精神刺激、发热、体位、运 动、前驱症状及与月经的关系等) 发作最初时的症状/体征(先兆、运动性表现等) 发作时表现(睁眼、闭眼、姿势、肌张力、运动症状、自主神经症状、自动症、知觉状态、舌咬伤、尿失禁等) 发作演变过程 发作持续时间 发作后表现(清醒、烦躁、嗜睡、朦胧状态、Todd麻痹、失语、遗忘、头痛、肌肉酸痛等) 发作频率和严重程度(包括持续状态史) 脑电图检查情况 其他辅助检查(血压、血糖、电解质、心电图、头部影像学等) 其他发作形式(如有,应按上述要点询问发作细节) 抗癫痫发作药使用情况(种类、剂量、疗程、疗效、不良反应、依从性等) 发作间期状态(精神症状、记忆力、焦虑、抑郁等) 发病后精神运动发育情况 既往史和家族史 围产史(早产、难产、缺氧窒息、产伤、颅内出血等) 中枢神经系统其他病史(感染、外伤、卒中、遗传代谢疾病等) 生长发育史(精神运动发育迟滞、倒退) 有无新生儿惊厥及热性惊厥史(简单型、复杂型) 家族史(癫痫、热性惊厥、偏头痛、睡眠障碍、遗传代谢疾病等) 疾病的影响 求学困难、失业、不能驾车、被过度保护、活动受限、心理压力等 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
(二)体格检查
应进行全身检查,但重点放在神经系统,包括:意识状态、认知状态、精神状态、局灶体征(偏瘫/偏盲等)、各种反射及病理征等。应注意观察头颅形状和大小、外貌、体重、身体畸形及排查某些神经皮肤综合征。体格检查对癫痫病因诊断有初步提示作用。有些体征则可能提示抗癫痫发作药的不良反应。
(三)辅助检查
1.脑电图(EEG)
癫痫发作最本质的特征是脑神经元异常过度放电,而EEG是能够反映脑电活动最直观、便捷的检查方法,是诊断癫痫发作、确定发作和癫痫的类型最重要的辅助手段,为癫痫患者的常规检查。当然,临床应用中也必须充分了解EEG(尤其头皮EEG)检查的局限性,必要时可延长监测时间或多次检查。
2.神经影像学磁共振成像(MRI)
对于发现脑部结构性异常有很高的价值。如果有条件,建议进行头颅MRI检查。头颅CT检查在显示钙化性或出血性病变时较MRI有优势。某些情况下,当临床已确诊为典型的特发性癫痫综合征(如儿童良性局灶性癫痫)时,可以不进行影像学检查。其他影像学检查,如功能磁共振成像(fMRI)、磁共振波谱(MRS)、单光子发射计算机断层成像(SPECT)、正电子发射断层成像(PET)等,均不是癫痫患者的常规检查。应注意,影像学发现的病灶与癫痫发作之间不一定存在必然的因果关系。
3.其他辅助检查
应根据患者具体情况进行选择。
(1)血液检查:
包括血常规、血糖、电解质、肝肾功能、血气、丙酮酸、乳酸、抗体等方面的检查,能够帮助查找病因。定期检查血常规、肝肾功能及电解质水平等指标还可辅助监测药物的不良反应。临床怀疑中毒时,应进行毒物筛查。已经服用抗癫痫发作药者,可酌情进行药物浓度监测。
(2)尿液检查:
包括尿常规及遗传代谢病的筛查。
(3)脑脊液检查:
主要排除颅内感染或免疫性炎性疾病,对某些遗传代谢病的诊断也有帮助。
(4)心电图:
对于疑诊癫痫或新诊断的癫痫患者,多主张常规进行心电图检查。这有助于发现容易误诊为癫痫发作的某些心源性发作(如心律失常所致的晕厥发作),还能早期发现某些心律失常(如长QT综合征、Brugada综合征和传导阻滞等),从而避免因使用某些抗癫痫发作药而可能导致的严重后果。
(5)遗传学检测:
临床疑诊癫痫的病因可能与遗传因素相关,可进行遗传学检测,分为下列情况进行:
1)一代测序(Sanger测序法):
临床诊断明确的特征性很强的癫痫综合征,且单一基因突变可以解释绝大多数患者(>80%),可以用一代Sanger测序法直接进行致病基因检测,例如Dravet综合征,80%以上是SCN1A基因的突变。如果上述均阴性,再进行二代测序。
2)二代测序遗传检测:
包括癫痫靶向基因包(Panel)、全外显子组(WES)、全基因组(WGS)检测。临床诊断无明显特异性特征的遗传性癫痫,有多个已知的致病基因:如婴儿痉挛症、伦诺克斯-加斯托综合征(Lennox-Gastaut syndrome)、发育性癫痫性脑病等,建议首选二代测序遗传检测,如果阴性,建议行染色体芯片(CMA)检测。
3)染色体芯片(CMA)检测:
该方法可发现基因组DNA拷贝数变异(copy number variation,CNV)。在癫痫发生之前即存在重度神经发育性疾病(智力障碍/发育迟缓,孤独症谱系疾病等)以及多发小畸形等情况下,可首先进行CMA检测,但是需要注意的是,有些染色体病相关癫痫,例如环形染色体20,只能通过染色体核型分析进行诊断,而染色体芯片不能诊断这种染色体变异。
4)高通量测序检测CNV:
随着高通量测序成本的降低和分析方法的日渐成熟,二代测序方法被越来越多地用于CNV的检测。低倍全基因组测序也称为基因组拷贝数变异测序(copy number variation sequencing,CNVseq),CNVseq检测具有低成本、高通量、低DNA样本量需求等优势。对于CNVseq检测结果建议使用平行方法如定量PCR(quantitative PCR,qPCR)等实验手段进一步验证确认。
一、概述
1981年ILAE癫痫发作分类曾是世界范围内应用最为广泛的发作分类(附录1),影响至今。2010年ILAE分类工作报告对癫痫发作的概念进行了修订。2017年ILAE推出了最新版本的癫痫发作分类。另外,症状学发作分类(semiological seizure classif i cation,SSC)在某些癫痫中心使用得更多。
二、癫痫发作的分类
(一)1981年ILAE癫痫发作分类
以临床表现和EEG改变(发作间期及发作期)作为分类依据,将癫痫发作分为:
1.部分性发作 最初的临床发作表现和EEG改变提示“一侧大脑半球内的一组神经元首先受累”。按照有无意识障碍,将部分性发作进一步分为简单部分发作、复杂部分发作和继发全面性发作。
2.全面性发作 最初的临床发作表现及EEG改变提示“双侧大脑半球同时受累”。
3.不能分类的发作。
(二)2010年ILAE分类工作报告
保留了对发作的“两分法”(即局灶性和全面性),但建议把“部分性”(partial)改称为“局灶性(focal)”,并根据需要可对局灶性发作进行具体描述(参见描述发作症状的术语,附录2)。较重要的是,此分类报告对癫痫发作的概念进行了修订:
1.局灶性发作
发作恒定地起源于一侧大脑半球内的、呈局限性或更广泛分布的致痫网络,并有着放电的优势传导途径,可以继发累及对侧半球。局灶性发作可以起源于皮质下结构。某些患者可以有多个致痫网络和多种发作类型,但每种发作类型的起始部位是恒定的。
2.全面性发作
发作起源于双侧大脑皮质及皮质下结构所构成的致痫网络中的某一点,并快速波及整个网络。每次发作起源点在网络中的位置均不固定。全面性发作时整个皮质未必均被累及,发作可不对称。
(三)2017年ILAE癫痫发作分类
为适应近30年来癫痫领域的长足发展和新的认识,ILAE于2017年推出最新版本的癫痫发作分类(表2-2)。新分类的框架在本质上仍沿用传统的“两分法”,在方法上仍主要基于症状学描述,但同时鼓励结合其他辅助检查资料(发作录像、脑电图及影像学等)来进行分类。新的发作分类是一种基于临床的实用性分类,强调可根据临床需求来选择分类的具体细化程度(基本版/扩展版)。
表2-2 2017年ILAE癫痫发作分类(扩展版)

| 局灶起始 全面性起始 起始不明 |
| 知觉保留 知觉障碍 运动症状 强直-阵挛 阵挛 强直 肌阵挛 肌阵挛-强直-阵挛 肌阵挛-失张力 失张力 癫痫性痉挛 非运动症状(失神) 典型失神 不典型失神 肌阵挛失神 眼睑肌阵挛失神 运动症状 强直-阵挛 癫痫性痉挛 非运动症状 行为中止 不能归类 运动症状起始 自动症 失张力 阵挛 癫痫性痉挛 过度运动 肌阵挛 强直 非运动症状起始 自主神经性 行为中止 认知性 情感性 感觉性 局灶进展为双侧强直-阵挛 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
与1981年分类相比,新版分类主要变化和特点包括:
1.重视发作的起始症状并将其作为细化分类的主要依据。无论局灶性还是全面性起始,发作均可大致分为运动和非运动症状两大类别。
2.增加了“起始不明”选项。对于有关发作起始信息不足的患者可暂时分为此类,可待日后信息完善时再确定局灶或全面起始。
3.在描述局灶性发作的意识状态用词方面,以简单易懂的“知觉(awareness)”取代较为复杂的“意识(consciousness)”。如,既往描述的“复杂部分性发作”可改为“知觉障碍性局灶性发作”。
4.新增了局灶性发作类型—失张力发作、阵挛发作、癫痫性痉挛、肌阵挛发作和强直发作,认为这些发作也可局灶性起源。另外,也增加了临床常见或可能具有定位提示意义的发作类型:自动症、过度运动性发作、行为中止性发作等。
5.对于局灶性发作建议废弃某些以往使用的术语,如认知障碍(dyscognitive)、简单部分(simple partial)、复杂部分(complex partial)及精神性(psychic)等。
6.新增了全面性发作类型—肌阵挛-强直-阵挛、肌阵挛-失张力、癫痫性痉挛、肌阵挛失神及眼睑肌阵挛失神。
7.以“局灶进展到双侧强直-阵挛发作(focal to bilateral tonic-clonic)”取代以前的“继发全面化(secondarily generalized)”。
(四)症状学发作分类(SSC)
从20世纪80年代开始,SSC在国外某些癫痫中心开始使用。SSC的特点可总结为:①舍弃了ILAE倡导的神经电生理检查参与发作分类的做法,SSC强调仅根据临床症状学进行发作分类(附录3);②为反映一次发作的主要演变,强调将主要发作类型(一般3~4种)按照时间顺序依次列出,并提供必要的被累及的躯体定位信息、意识状况或定侧体征。举例:腹部先兆→左手自动运动发作(意识丧失)→全面强直-阵挛发作,定侧体征:右手肌张力障碍姿势。SSC在某些以术前评估为工作重点的中心更受青睐。
三、常见癫痫发作类型及诊断要点
根据2017年ILAE癫痫发作分类,具体发作类型及诊断要点描述如下:
(一)全面性发作(generalized seizures)
1.全面性强直-阵挛发作(generalized tonic-clonic seizure,GTCS)是一种表现最明显的发作形式,故既往也称为大发作(grand mal)。以意识丧失、双侧对称强直后紧跟有阵挛动作并通常伴有自主神经受累表现为主要临床特征。
2.强直发作(tonic seizure)表现为躯体中轴、双侧肢体近端或全身肌肉持续性的收缩、肌肉僵直。通常持续2~10秒,偶尔可达数分钟。发作时EEG显示双侧性波幅渐增的棘波节律([20±5)Hz]或低波幅(约10Hz)节律性放电活动。强直发作是Lennox-Gastaut综合征的最主要发作类型。
3.阵挛发作(clonic seizure)表现为双侧肢体节律性(1~3Hz)的抽动,伴有或不伴有意识障碍,多持续数分钟。发作时EEG为全面性(多)棘波或(多)棘-慢波综合。
4.肌阵挛发作(myoclonic seizure)表现为不自主、快速短暂、电击样肌肉抽动,每次抽动历时10~50毫秒,很少超过100毫秒。可累及全身也可限于某局部肌肉或肌群。可非节律性反复出现。发作期典型的EEG表现为暴发性全面性多棘-慢波综合。肌阵挛发作既可见于一些预后较好的特发性癫痫患者(如青少年肌阵挛性癫痫),也可见于一些预后较差的、有弥漫性脑损害的癫痫性脑病(如Dravet综合征、Lennox-Gastaut综合征)。
5.失张力发作(atonic seizure)表现为头部、躯干或肢体肌肉张力突然丧失或减低,发作之前没有明显的肌阵挛或强直成分。发作持续约1~2秒或更长。临床表现轻重不一,轻者可仅有点头动作,重者可导致站立时突然跌倒。发作时EEG表现为短暂全面性2~3Hz(多)棘-慢波综合发放或突然电压减低。失张力发作多见于癫痫性脑病[如Lennox-Gastaut综合征、多泽综合征(Doose syndrome)]。
6.肌阵挛-强直-阵挛发作(myoclonic-tonic-clonic seizure)表现双侧肢体单次或数次阵挛或肌阵挛性抽动,随后演变为强直-阵挛性发作。这种发作类型多见于青少年肌阵挛性癫痫。
7.肌阵挛-失张力发作(myoclonic-atonic seizure)一种表现为肢体或躯干先出现肌阵挛性抽动,随后出现肌张力降低的发作类型,立位时发作可能导致患者跌倒。曾称为“肌阵挛-站立不能性发作(myoclonic-astatic seizure)”。这种发作常见于Doose综合征。
8.失神发作(absence seizures)
(1)典型失神(typical absence):发作突发突止,表现为动作突然中止或明显变慢,意识障碍,不伴有或伴有轻微的运动症状(如阵挛/肌阵挛/强直/自动症等)。发作通常持续5~20秒(<30秒)。发作时EEG呈双侧对称同步、3Hz(2.5~4Hz)的棘-慢综合波暴发。约90%的典型失神患者可被过度换气诱发。主要见于儿童和青少年,如儿童失神癫痫和青少年失神癫痫,罕见于成人。
(2)不典型失神(atypical absence):发作起始和结束均较典型失神缓慢,意识障碍程度较轻,伴随的运动症状(如自动症)也较复杂,肌张力通常减低,发作持续可能超过20秒。发作时EEG表现为慢的(<2.5Hz)棘-慢综合波节律。主要见于严重神经精神障碍的患者,如Lennox-Gastaut综合征。
(3)肌阵挛失神(myoclonic absence):表现为失神发作的同时,出现肢体节律性2.5~4.5Hz肌阵挛性动作,并伴有强直成分。发作期EEG与典型失神类似。主要见于肌阵挛失神癫痫。
(4)眼睑肌阵挛失神(absence with eyelid myoclonia):表现为失神发作的同时,眼睑和/或前额部肌肉出现5~6Hz肌阵挛动作。发作期EEG显示全面性3~6Hz多棘-慢综合波。常见于Jeavons综合征。
(二)局灶性发作(focal seizures)
1.知觉保留/知觉障碍的局灶性发作(focal aware or focal impaired awareness seizures)分别用来描述发作时知觉有保留或知觉有障碍的局灶性发作。此处“知觉(awareness)”被定义为“感知自我和环境”。如果不能明确局灶性发作时知觉状态,则可简单地描述为“局灶性发作”。
2.自动症(automatisms)指的是通常在知觉障碍状态下,患者作出的反复刻板、无目的或似乎有目的、基本协调的不自主动作或行为。常见自动症类型包括口咽自动症、手部自动症、言语性自动症及过度运动性自动症等。
3.过度运动性发作(hyperkinetic seizure)是一种主要累及躯干及肢体的近端,动作幅度通常较大、快速剧烈的局灶性运动性发作。例如,上肢快速挥舞样运动或下肢反复蹬踏样动作。
4.自主神经性发作(autonomic seizure)指发作时以自主神经功能发生明显改变为主要表现的非运动局灶性发作。自主神经改变可能涉及心肺、瞳孔、胃肠、泌汗、血管舒缩和体温调节等功能,常被描述为心动过速、过度换气、胃气上升、脸红、面色苍白、恶心呕吐及竖毛等。
5.行为中止性发作(behavior arrest)指从发作起始就以动作行为中止为主要表现并贯穿整个发作过程的非运动局灶性发作。
6.认知性发作(cognitive seizure)以语言、思维或其他高级皮质功能改变为主要表现的非运动局灶性发作。例如,似曾相识感(déjà vu)、幻觉或错觉性发作、失语性发作及强迫思维发作等。
7.情感性发作(emotional seizure)以情绪改变为主要表现的非运动局灶性发作。例如,发作性恐惧(害怕)、焦虑、生气、激越、高兴、欣快等。
8.感觉性发作(sensory seizures)指的是非外源性刺激诱发的自我感知体验性发作。临床常见类型包括躯体感觉性、视觉性、听觉性、嗅觉性、味觉性、温度觉性或前庭性发作等。
9.局灶进展为双侧强直-阵挛发作(focal to bilateral tonic-clonic seizure)是一种局灶起源的运动性或非运动性发作,进而发展为双侧强直-阵挛性发作。该发作类型本质上仍为局灶性发作,既往曾被描述为部分发作继发全面化(partial seizure with secondary generalization)。
通常情况下,以上各种局灶性发作的发作期EEG表现为局灶起始、有演变特征的痫性活动,具体表现形式可因放电的起始部位、扩散速度和范围等因素的不同而各异。
(三)癫痫性痉挛(epileptic spasms)
最初在2010年ILAE分类工作报告中明确提出将癫痫性痉挛作为一种发作类型。癫痫性痉挛可以是全面性起源、局灶性起源或起源不明。
癫痫性痉挛表现为突然、主要累及躯干中轴和双侧肢体近端肌肉的强直性收缩,历时0.2~2秒,突发突止。临床可分为屈曲型或伸展型痉挛,以前者多见,表现为发作性点头动作,常在觉醒后成串发作。发作间期EEG表现为高度失律或类高度失律,发作期EEG表现多样化(电压降低、高幅双相慢波或棘慢波等)。癫痫性痉挛多见于婴幼儿,如West综合征,也可见于其他年龄。
(四)反射性发作(reflex seizures)
反射性发作不是独立的发作类型。它既可以表现为局灶性发作,也可以为全面性发作。其特殊之处是,发作具有特殊的外源性或内源性促发因素,即每次发作均为某种特定感觉刺激所促发,并且发作与促发因素之间有密切的锁时关系。促发因素包括视觉、思考、音乐、阅读、进食、操作等非病理性因素。可以是简单的感觉刺激(如闪光),也可以是复杂的智能活动(如阅读、下棋)。发热、酒精或药物戒断等病理性情况下诱发的发作不属于反射性发作。反射性发作和自发性发作可同时出现在一个癫痫患者中。
四、新生儿癫痫发作的诊断及分类
上述有关癫痫发作分类的内容主要是针对成人和大龄儿童,并不适合新生儿。鉴于新生儿癫痫发作在临床及电生理表现、病因学、诊断方法等方面的差异,2021年ILAE发布了新生儿癫痫发作分类。此分类的总体框架与2017版分类大体一致,可认为是后者的新生儿修订版。
(一)新生儿癫痫发作的诊断和分类
强调脑电图是诊断新生儿癫痫发作的“金标准”,即可以仅靠脑电图表现来诊断。如果只有临床事件而无相应的脑电图发作图形,则不能诊断为癫痫发作。新生儿癫痫发作可分为脑电图-临床发作和脑电图发作。对于前者,可根据具体表现进一步确定发作类型:运动性发作(自动症、阵挛、癫痫性痉挛、强直)和非运动性发作(自主神经性、行为中止)。要注意的是,新生儿只有局灶性发作,不存在全面性发作。
新生儿癫痫发作的分类及诊断流程见附录4。
(二)新生儿癫痫发作的诊断级别
鉴于具体诊断手段可能存在差异(持续脑电图监测、振幅整合脑电图或仅靠肉眼观察),新生儿癫痫发作的诊断可靠性存在差异。新生儿癫痫发作的诊断级别见附录5。
1989年ILAE推出《癫痫和癫痫综合征的国际分类》方案,鉴于近二十余年来陆续发现了一些新的癫痫综合征类型,以及对癫痫及癫痫综合征病因学的深入研究,ILAE一直在尝试对癫痫及癫痫综合征相关术语进行修订和补充,以期建立一个更为完善的分类系统,并于2017年提出了癫痫的最新分类框架。
一、1989年ILAE癫痫及癫痫综合征分类
1989年ILAE将癫痫及癫痫综合征分为四大类:部位相关性(局灶性、局限性、部分性)癫痫及综合征、全面性癫痫及综合征、不能确定为局灶性还是全面性的癫痫及综合征、特殊综合征,并从病因学的角度,将癫痫及癫痫综合征主要分为三种类型(附录6):①特发性癫痫及综合征:定义为除了可能的遗传易感性之外,没有其他潜在的病因。患者除了癫痫发作之外,没有结构性脑部病变和其他神经系统症状或体征。癫痫发作通常有年龄相关性,如儿童失神癫痫、青少年肌阵挛癫痫。②症状性癫痫及综合征:定义为癫痫发作是由一个或多个可辨认的结构性脑部病变引起。如海马硬化引起的内侧颞叶癫痫、局灶性皮质发育不良引起的额叶癫痫。③隐源性癫痫及综合征:推测病因也是症状性的,但目前的检查手段无法明确病因。随着高分辨率MRI的应用以及遗传病因学的进展,隐源性癫痫的数量将越来越少。
二、2010年ILAE关于癫痫及癫痫综合征分类的修订
2010年ILAE提出了癫痫的过渡性分类框架,将癫痫的病因分为结构性、遗传性/代谢性、病因不明性,并以起病年龄对癫痫综合征进行了分组(附录7),包括新生儿期、婴儿期、儿童期、青少年-成年期及起病年龄可变的癫痫综合征。2010年的分类为2017年癫痫分类框架的修订奠定了基础。
三、2017年ILAE癫痫分类框架
2017年ILAE分类与命名委员会更新了癫痫的分类框架(图2-1),以反映科学取得巨大进步后对癫痫及其基本机制认识的进步。2017年的癫痫分类框架已在临床得到广泛应用,此分类呈现了三个层次,首先是明确发作类型;明确发作类型后,下一步是诊断癫痫类型,包括局灶性癫痫、全面性癫痫、全面性及局灶性癫痫两者兼有,以及分类不明的癫痫;第三层次是癫痫综合征,此处可以作出特定综合征的诊断。这一新分类强调在每一步诊断时都要考虑癫痫的病因,因为病因将会对治疗产生重要影响。新分类将癫痫病因分为6个亚组(结构性、遗传性、感染性、代谢性、免疫性、未知)是基于其潜在的治疗因果关系,本章第五节将详细叙述病因分类。2017年的癫痫分类框架同时强调应关注癫痫共患病的诊断,如注意缺陷多动障碍、孤独症谱系障碍等,有助于提高癫痫患者的综合管理水平。
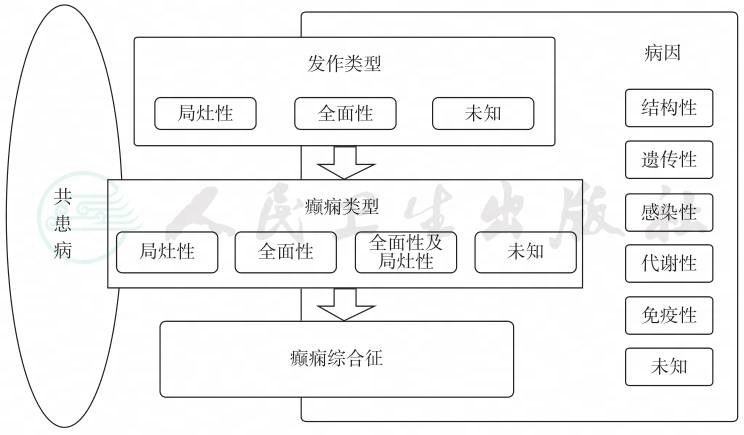
图2-1 2017年ILAE癫痫分类框架
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
随着对癫痫发作和癫痫神经生物学认识的进步,关于分类基本概念的主要术语已经发生转变,2017年的分类引入了新的术语,如“发育性癫痫性脑病(developmental and epileptic encephalopathy)”是指癫痫患儿出现的脑病与病因及癫痫活动均相关,即使癫痫发作能够完全控制,其脑病表现也不能完全恢复,甚至还可能随着年龄增长而继续加重。“自限性(selflimited)”和“药物有效性(drug-responsive)”取代“良性(benign)”一词。癫痫分类的进展旨在反映当前对癫痫的理解,使其与临床实践密切结合,并能成为临床和科研领域交流的基础工具。
四、2022年ILAE癫痫综合征分类
2022年4月ILAE疾病分类与定义工作组发布了癫痫综合征新的分类方案,将癫痫综合征分为以下四组(附录8):①新生儿期和婴儿期起病的癫痫综合征(epilepsy syndromes in the neonate and infant),发病年龄<2岁;②儿童期起病的癫痫综合征(epilepsy syndromes with onset in childhood),发病年龄2~12岁;③起病年龄可变的癫痫综合征(epilepsy syndromes with onset at a variable age),儿童和成年期均可发病;④特发性全面性癫痫综合征(idiopathic generalized epilepsysyndromes)。新的癫痫综合征分类方案先依据起病年龄将癫痫综合征分组,再结合发作类型、病程和病因将癫痫综合征归类。如新生儿期和婴儿期起病的癫痫综合征可进一步分为自限性癫痫(如自限性新生儿癫痫、自限性婴儿癫痫等)、发育性癫痫性脑病(如婴儿癫痫伴游走性局灶性发作、葡萄糖转运子1缺陷综合征、Dravet综合征等)和病因特异性癫痫性脑病(如KCNQ2-发育性癫痫性脑病、原钙黏蛋白19簇集性癫痫等)。2022年癫痫综合征新的分类方案刚刚发布,其实用性有待临床进一步检验,并在以后的分类更新中不断完善。
五、常见癫痫综合征的临床特点(2010年ILAE分类)
(一)良性家族性新生儿癫痫
良性家族性新生儿癫痫(benign familial neonatal epilepsy,BFNE)既往又称良性家族性新生儿惊厥,是一种少见的常染色体显性遗传性疾病。致病基因包括KCNQ2和KCNQ3,以KCNQ2变异更常见。KCNQ2定位于染色体20q13.33,编码电压门控钾离子通道KQT样亚家族成员2。本病的主要特点是正常足月新生儿出生后不久(多数在7天内)出现强直、阵挛性惊厥发作,常合并自主神经症状和运动性自动症,发作频繁、短暂。发作间期患儿一般状态良好,除家族中有类似发作史和脑电图非特异性改变之外,其他病史和检查均正常。预后良好,惊厥发作多于2~4周内消失。EEG发作间期大多正常,部分病例有全面性或局灶性异常。
(二)良性家族性婴儿癫痫
良性家族性婴儿癫痫(benign familial infantile epilepsy,BFIE)既往又称良性家族性婴儿惊厥,为常染色体显性遗传,可有外显率不全。约60%~80%的家系可发现致病基因,包括PRRT2、KCNQ2、SCN2A和SCN8A,以PRRT2基因最常见,该基因编码富脯氨酸跨膜蛋白。本病起病年龄为3~20个月,绝大多数在1岁以内发病,起病前后智力运动发育正常,表现为局灶性发作或局灶性发作继发全面性发作,发作常呈丛集性,无癫痫持续状态。EEG发作间期背景正常,无典型癫痫样放电,睡眠期可有Rolandic区小棘慢波;发作期EEG放电可起源于颞区、顶区、枕区或额区。头颅影像学检查无异常,诊断时要排除低钙血症、低血糖等代谢紊乱导致的惊厥。本病对抗癫痫发作药效果好,预后良好,2岁后不再发作。PRRT2变异的家系部分受累者在儿童期或青少年可出现阵发性运动诱发的运动障碍(paroxysmal kinesigenic dyskinesias,PKD),这种BFIE的临床亚型被称为婴儿惊厥伴阵发性舞蹈手足徐动症(infantile convulsions with paroxysmal choreoathetosis syndrome,ICCA)。
(三)良性婴儿癫痫
良性婴儿癫痫(benign infantile epilepsy,BIE)早期又称良性婴儿惊厥,发病年龄为生后3~20个月,病因主要与遗传易感性有关,少数患儿可发现有PRRT2、KCNQ2、SCN2A或SCN8A基因变异。其临床和脑电图特点与良性家族性婴儿癫痫受累者相似,只是没有良性婴儿癫痫家族史,为散发病例。预后良好,2岁后可自行缓解。
(四)大田原综合征
大田原综合征(Ohtahara syndrome)被认为是癫痫性脑病发病最早的形式,由日本学者大田原(Ohtahara)于1977年首次报道。多数患儿有严重的先天性脑发育异常或围产期脑损伤。没有发现明确病因的患儿中,通过二代测序的方法发现少数患儿可由致病基因STXBP1、ARX、PLCB1、PNKP、SCN2A、KCNQ2、GNAO1等变异导致。起病年龄在生后3个月之内,多数可早到生后1个月内发病,表现为强直痉挛发作,脑电图特点为暴发-抑制图形,有严重的精神运动发育落后,发作难以控制,预后极差,死亡率高,存活者可演变为West综合征和Lennox-Gastaut综合征。
(五)早期肌阵挛性脑病
早期肌阵挛性脑病(early myoclonic encephalopathy,EME)与大田原综合征有某些共同特点,如婴儿早期起病及脑电图暴发-抑制图形。主要区别点在于病因和发作类型不同。病因多不清楚,有些病例为先天代谢性障碍,如丙酸血症、非酮症性高甘氨酸血症等。近年来发现部分病例由致病基因SLC25A22或PNPO变异导致。其临床特点为生后3个月内发病,可早到新生儿期发病。出现节段性、游走性肌阵挛,主要累及四肢远端及面部小肌群。以后有频繁的局灶性发作,部分患者有肌阵挛和强直痉挛发作。脑电图表现为暴发-抑制图形,睡眠期明显。病情严重,死亡率高,存活者常有精神运动发育迟滞,预后差,属于发育性癫痫性脑病。
(六)婴儿癫痫伴游走性局灶性发作
婴儿癫痫伴游走性局灶性发作(epilepsy of infancy with migrating focal seizures,EIMFS)早期又称为婴儿游走性部分性癫痫,是一种罕见的婴儿早期发病的癫痫性脑病,多数为散发病例,少数可有家族史。其病因主要与遗传因素有关,可由致病基因KCNT1、SCN1A、SCN2A或SCN8A杂合新生变异导致,呈常染色体显性遗传。也可由致病基因PLCB1、SLC25A22、BRAT1、TBC1D24或SLC12A5复合杂合变异导致,呈常染色体隐性遗传。EIMFS的临床特点为生后3个月内起病,可早到新生儿期发病,发作表现为游走性局灶性发作,发作在一侧半球内或双侧半球之间游走,发作频率逐渐增多,最终发展为持续性发作并伴有发育倒退。脑电图发作间期为大量多灶性放电,发作期为游走性多灶性放电。本病对抗癫痫发作药疗效差,预后不良,患儿可死于癫痫持续状态。
(七)Dravet综合征
Dravet综合征(Dravet syndrome)又称婴儿严重肌阵挛癫痫(severe myoclonic epilepsy in infancy),因发现少数患儿病程中可始终不出现肌阵挛发作,2001年ILAE将其更名为Dravet综合征。本病由法国医生Charlotte Dravet于1978年首先报道,本病多为散发病例,少数有热性惊厥或癫痫家族史。约80%的患儿可发现钠离子通道基因SCN1A变异,多数为新生变异,少数为遗传性变异。本病少数患儿由致病基因PCDH19、SCN2A、SCN8A、SCN1B、GABRA1、GABRB2、GABRG2、CHD2、ALDH7A1、HCN1 或KCNA2 变异导致。其临床特点为1岁以内起病,生后6个月为高峰发病年龄,首次发作多表现为热性惊厥,1岁以内主要表现为发热诱发的持续时间较长的全面性或半侧阵挛发作,1岁后逐渐出现多种形式的无热发作,包括全面性强直-阵挛发作、半侧阵挛发作、肌阵挛发作、不典型失神、局灶性发作,发作具有热敏感的特点,易发生癫痫持续状态,约30%的患儿发作有光敏感的特点。早期发育正常,1岁后逐渐出现智力运动发育落后或倒退,约60%的患儿可出现共济失调。脑电图在1岁以前常正常,1岁以后出现广泛性棘慢波、多棘慢波或局灶性、多灶性痫样放电。多数患儿对抗癫痫发作药疗效差,成年期仍有发作,智力发育落后,预后不良,属于发育性癫痫性脑病。本病死亡率高,文献报道可达10%,可由于癫痫猝死或发热诱发的严重癫痫持续状态导致急性脑病死亡。
(八)婴儿痉挛症
婴儿痉挛症(infantile spasms)又称West综合征,由West医生于1841年首次报道。病因复杂多样,可由先天性脑发育异常、遗传代谢病、围产期脑损伤、中枢神经系统感染后脑损伤等导致。近年来发现少数患儿可由致病基因STXBP1、ARX、CDKL5、FOXG1、IQSEC2、TSC1、TSC2、MAGI2、SPTNA1、SCN2A、GRIN2B、DNM1、PLCB1、ST3GAL3、PIGA、SLC35A2和DOCK7等变异导致。通常生后3~12个月发病,很少在3个月内或1岁后发病。特征性表现为癫痫性痉挛发作、脑电图显示高度失律和精神运动发育落后三联征。本病为临床最常见的癫痫性脑病,多数患儿治疗效果不佳,预后不良,部分可演变为Lennox-Gastaut综合征。
(九)婴儿肌阵挛癫痫
婴儿肌阵挛癫痫(myoclonic epilepsy in infancy)早期称为良性婴儿肌阵挛癫痫,是一种临床少见的癫痫综合征,病因可能与遗传易感性有关。其主要特点为1~3岁发病,表现全面性肌阵挛发作,不伴其他发作类型。发作期脑电图为广泛性棘慢波或多棘慢综合波。发病前发育正常,多数患儿发作易于控制,发作可在4~11岁缓解,预后良好。少数患儿发作难以控制,可遗留认知损害。
(十)Lennox-Gastaut综合征
Lennox-Gastaut综合征(Lennox-Gastaut syndrome,LGS)是一种临床常见的年龄相关性癫痫性脑病,1939年由Lennox首先报道其临床和脑电图特点,1966年由Gastaut加以补充。LGS部分病例可由West综合征演变而来。病因复杂多样,包括脑发育异常、围产期脑损伤、中枢神经系统感染或外伤等导致的脑损伤。近年来发现少数病例可由致病基因CHD2、SCN2A、SCN8A、GRIN2B、ALG13、GABRB3、STXBP1和 MT-ND1变异导致。多发生于1~8岁儿童,主要特点为多种癫痫发作类型、脑电图显示广泛性慢的(1.5~2.5Hz)棘慢综合波和智力发育落后三联征。最常见的发作类型有强直发作、不典型失神及失张力发作,也可有肌阵挛、全面强直-阵挛和局灶性发作。通常发作频繁,药物难以控制,总体预后不良。
(十一)肌阵挛-失张力癫痫
肌阵挛-失张力癫痫(myoclonic-atonic epilepsy,MAE)又称Doose综合征,由德国医生Herman Doose于1970年首次报道,临床相对少见。病因不明,可能与遗传易感性有关,近年来发现 SCN1A、SCN2A、SCN1B、STX1B、GABRG2、SLC2A1、CHD2、SYNGAP1、NEXMIF、KIAA2022和SLC6A1基因变异可导致Doose综合征表型。发病年龄1~5岁,发病急剧,常以全面性强直-阵挛发作起病,很快出现多种形式的全面性发作,包括肌阵挛、肌阵挛-失张力、失张力和不典型失神发作,发作频繁,可由于肌阵挛、肌阵挛-失张力或失张力发作导致跌倒,部分患儿可出现不典型失神持续状态。脑电图显示广泛性不规则的2.5~3Hz(多)棘慢综合波,肌阵挛-失张力或失张力发作时同步肌电图可见短暂电静息期。本病预后变化大,约70%的患者对抗癫痫发作药治疗有效,发作最终可缓解,预后良好。少数患者病程后期可出现强直发作,进展为Lennox-Gastaut综合征。多数患者智力正常或接近正常,少数发作不能及时控制的患者,出现智力发育落后。
(十二)儿童失神癫痫
儿童失神癫痫(childhood absence epilepsy,CAE)是儿童期常见的特发性全面性癫痫(idiopathic generalized epilepsy,IGE),单卵双胎共患率显著高于双卵双胎,支持发病与遗传因素有关,已发现 CACNA1H、GABRA1、GABRB2、GABRB3、GABRG2、GABRD 和 SLC2A1基因是其易感基因。起病年龄4~10岁,临床表现为频繁的典型失神发作。脑电图背景正常,发作期为双侧广泛、同步、对称性3Hz棘慢波节律。患儿发育正常,对抗癫痫发作药效果好,常在12岁前缓解,预后良好。
(十三)眼睑肌阵挛癫痫
眼睑肌阵挛癫痫(eyelid myoclonic epilepsy,EME)又称Jeavons综合征,是一种儿童期起病的遗传性全面性癫痫,1977年由Jeavons首次提出,病因与遗传易感性有关,已明确的致病基因包括SYNGAP1、CHD2、RORB和NEXMIF。起病高峰年龄2~14岁,以眼睑肌阵挛为最突出表现,持续时间较长时可伴失神,发作频繁,每日可达数十次,持续时间3~6秒。具有反射性发作特征,在明亮环境下合眼诱发,对间断闪光刺激(IPS)或其他闪烁光敏感。病程中可出现全面性强直-阵挛发作。发作间期EEG显示广泛性放电,有时后头部(枕区)限局;发作期EEG为广泛对称同步3~6Hz棘慢波、多棘慢波暴发,持续0.5至数秒,持续超过4秒以上常伴有失神发作,合眼及IPS均诱发放电伴发作,同步肌电图显示临床发作伴同步眼睑肌电暴发。本病为终生性疾病,发作控制困难或停药困难。改善生活方式及避免诱发因素对本病的治疗也非常重要。发作难以控制、病程长者对认知、行为有影响。
(十四)肌阵挛失神癫痫
肌阵挛失神癫痫(epilepsy with myoclonic absences)是一种少见的儿童癫痫综合征,20%有癫痫家族史(多数为全面性癫痫)。2/3的病例未找到明确病因,可能与遗传易感性有关。1/3的病例有围产期脑损伤、脑发育异常、染色体异常[12p三体综合征、安格尔曼综合征(Angelman syndrome)]等,少数患儿有智力损害或头颅影像学异常。起病高峰年龄7岁(11个月~12.5岁),以肌阵挛失神为主要发作类型,部分患者还可出现全面性强直-阵挛发作或失张力发作。发作间期EEG背景活动正常或轻度非特异性异常,可见广泛性棘慢波散发或短阵暴发,14%的病例IPS可诱发发作;发作期EEG为双侧对称同步的3Hz棘慢波节律暴发,棘慢波的频率与肌阵挛的频率相同,同步肌电图对鉴别典型失神发作和肌阵挛失神发作有帮助。药物治疗反应欠佳,总体预后不如儿童失神癫痫好。
(十五)儿童良性癫痫伴中央颞区棘波
儿童良性癫痫伴中央颞区棘波(benign epilepsy in childhood with centrotemporal spikes,BECTS)又称为良性Rolandic癫痫,是儿童期最常见的癫痫综合征,发病有明显的年龄依赖性,病因可能与遗传易感性有关。多数患者5~10岁发病(3~12岁)。主要特点是面部和口咽部局灶运动性和感觉性发作,可继发全面性发作。大多数患儿仅在睡眠中发作,通常发作不频繁。EEG的特征为中央颞区棘波,在睡眠中发放明显增多,对抗癫痫发作药疗效好,几乎所有病例在16岁前缓解,预后良好。
值得注意的是,少数早期诊断为BECTS的患儿,在随访中可演变为BECTS变异型。BECTS变异型的特点包括:①病程早期符合BECTS的临床特点;②病程中出现新的发作类型(负性肌阵挛、不典型失神)和/或口咽部运动障碍;③EEG显示Rolandic区限局性放电在清醒期及睡眠期均明显增多,符合睡眠中癫痫性电持续状态(electrical status epilepticus during sleep,ESES)的诊断标准;④起病后可出现轻度的认知损伤。
当BECTS患儿在随访的过程中出现以下情况时应警惕演变为BECTS变异型的可能:①睡眠中局灶性发作加重;②日间出现新的发作类型;③EEG显示清醒期Rolandic区放电明显增多,NREM睡眠期棘慢波指数大于50%以上。早期明确诊断BECTS变异型,有利于制定正确的治疗策略,从而避免使用加重发作的药物。BECTS变异型属年龄依赖性的自限性疾病,癫痫发作缓解及EEG恢复正常的年龄与BECTS相似,但由于持续大量的痫样放电可遗留认知损伤,故总体长远预后不如典型BECTS好。
(十六)Panayiotopoulos综合征
Panayiotopoulos综合征(Panayiotopoulos syndrome)既往又称早发型儿童良性枕叶癫痫,2010年ILAE将其更名为Panayiotopoulos综合征,病因不明,年龄依赖性发病,可能与遗传易感性有关。绝大多数3~6岁发病(1~14岁),主要临床特征为以呕吐为主的自主神经症状性发作及自主神经发作持续状态,其他少见的自主神经症状为苍白、流涎、瞳孔放大等,多数病例可出现眼球偏斜,发作后期部分病例可出现半侧阵挛或进展为双侧强直-阵挛发作。脑电图显示枕区为主的多灶性棘波放电,约1/3的病例棘波发放可在枕区以外。对抗癫痫发作药疗效好,预后良好。
(十七)晚发型儿童枕叶癫痫(Gastaut型)
晚发型儿童枕叶癫痫(Gastaut型)发病较Panayiotopoulos综合征晚,发病年龄3~16岁,一般认为发病与遗传易感性有关。主要临床特点为发作性视幻觉或黑矇,日间发作为主,也可表现为发作性眼球偏斜、眼震、眼睑扑动、半侧阵挛或继发双侧强直-阵挛发作。脑电图显示枕区阵发性放电。对抗癫痫发作药疗效好,预后良好。
(十八)Landau-Kleffner综合征
Landau-Kleffner综合征(Landau-Kleffner syndrome,LKS)又称获得性癫痫性失语(acquired epileptic aphasia),于1957年由Landau和Kleffner首次报道。本病少见,病因不明,年龄依赖性发病,可能与遗传因素有关,文献报道少数患儿可发现GRIN2A、SETD1B基因变异。起病年龄多在2~8岁。临床特点为获得性失语、癫痫发作、脑电图异常和行为心理障碍。癫痫发作主要表现局灶性运动性发作,多在睡眠中出现。清醒时可出现不典型失神、肌阵挛或失张力发作。脑电图以慢波睡眠期连续出现的棘慢综合波为特征,多为双侧性,颞区为主。癫痫发作和脑电图改变呈年龄依赖性,常在15岁后缓解。半数以上患者持续有语言、心理和行为障碍。
(十九)癫痫性脑病伴慢波睡眠期持续棘慢波
癫痫性脑病伴慢波睡眠期持续棘慢波(epileptic encephalopathy with continuous spike and waves during slow wave sleep,CSWS)属于癫痫性脑病,为年龄依赖性发病,主要见于儿童期。1/3的患儿起病前即有神经系统异常,包括围产期脑损伤和先天性脑发育异常。2/3的患儿病因不明,约13%~15%的患儿有癫痫家族史,故遗传因素可能与部分患儿的发病有关,少数患儿可发现GRIN2A、FOXP1、KCNMA1和CSNK1D基因新生杂合变异。主要特征为多种类型的癫痫发作,包括局灶性发作、不典型失神、肌阵挛发作和失张力发作。脑电图在慢波睡眠期呈ESES状态,有神经心理和运动行为障碍。CSWS患儿的EEG清醒期可见一侧或双侧额区、中央区、Rolandic区或额、颞区为主的限局性棘波、棘慢波。睡眠期为广泛性持续的1.5~4.0Hz棘慢复合波发放,间有少量额区或额颞区为主的局灶性异常。ESES与神经心理损伤密切相关。本病2/3的患儿发病前智力运动发育正常,1/3有智力发育落后。少数患儿有痉挛性四肢瘫、偏瘫、共济失调等神经系统异常体征。CSWS患儿不论起病前的智力运动水平如何,起病后均会有新的损伤出现。多数患儿在EEG出现ESES期间,常表现为全面性的认知倒退。运动倒退也见于半数患儿,表现为精细动作差等共济失调、偏瘫。语言障碍在CSWS患儿中也可出现,但主要表现为语言表达障碍。CSWS患儿神经心理学损伤的程度和表现与ESES的严重程度、累及部位、持续时间等多种因素有关。CSWS患儿癫痫发作一般呈良性演变过程,在青春期前后消失,但患儿有广泛的认知障碍、智力倒退及行为问题。
(二十)青少年失神癫痫
青少年失神癫痫(juvenile absence epilepsy,JAE)是常见的IGE之一,病因不明,可能与遗传易感性有关,已发现GABRB2、GABRG2、GABRA1、CACNA1A和SLC2A1是其易感基因。发病年龄多在7~16岁,高峰为10~12岁。主要临床特征为典型失神发作,约80%的病例伴有全面性强直-阵挛发作,约15%的病例还有肌阵挛发作。发作期脑电图为双侧广泛同步、对称性3~4Hz棘-慢综合波节律,多数病例药物治疗后缓解,预后相对良好。
(二十一)青少年肌阵挛癫痫
青少年肌阵挛癫痫(juvenile myoclonic epilepsy,JME)为常见的IGE,病因不明,可能与遗传易感性有关,已发现GABRA1、GABRD、CACNB4、EFHC1和EFHC2是其易感基因。通常起病于12~18岁,生长发育及神经系统检查正常。临床主要表现为觉醒后不久出现肌阵挛发作,80%以上的病例有全面性强直-阵挛发作,约1/3的病例有失神发作。发作间期脑电图为广泛性4~6Hz多棘慢综合波。本病对抗癫痫发作药治疗反应好,但多数患者需长期治疗。
(二十二)仅有全面性强直-阵挛发作的癫痫
仅有全面性强直-阵挛发作的癫痫(epilepsy with generalized tonic-clonic seizures only)发病年龄为5~50岁,高峰年龄为10~20岁。本综合征包含了1989年ILAE提出的觉醒期强直-阵挛发作性癫痫(epilepsy with generalized tonic-clonic seizures on awakening),属于IGE。全部患者均有全面性强直-阵挛发作,可发生于任何时间(睡眠、清醒或觉醒时),无其他发作类型。脑电图特点为广泛性4~5Hz多棘慢综合波或多棘波发放。预后良好。
(二十三)遗传性癫痫伴热性惊厥附加症
遗传性癫痫伴热性惊厥附加症(genetic epilepsy with febrile seizures plus,GEFS+)既往又称全面性癫痫伴热性惊厥附加症(general epilepsy with febrile seizures plus,GEFS+),为家族性遗传性癫痫综合征。该综合征1997年由澳大利亚Scheffer医生首先报道。家系成员发病年龄主要在儿童期和青少年期。已报道的致病基因包括SCN1A、SCN1B、SCN2A、GABRG2、GABRD和STX1B。家系成员的临床表型具有异质性,最常见的表型为热性惊厥(febrile seizures,FS)和热性惊厥附加症(febrile seizures plus,FS+)、其次为 FS/FS+伴肌阵挛发作、FS/FS+伴失神发作、FS/FS+伴失张力发作、FS/FS+伴局灶性发作,其他少见的表型为局灶性癫痫、特发性全面性癫痫(如CAE、JAE、JME),个别患者表现为Dravet综合征或Doose综合征。家族成员中有FS和FS+病史是GEFS+家系诊断的重要依据。GEFS+家系受累成员的具体表型诊断要依据每个个体的发作类型和脑电图特点确定。GEFS+家系受累者总体预后良好,青春期后不再发作,但如果为Dravet综合征,则预后不良。
(二十四)进行性肌阵挛癫痫
进行性肌阵挛癫痫(progressive myoclonus epilepsies,PME)的概念于1903年由Herman Lundborg首先提出,病因包括一组神经遗传代谢病,大多数为家族遗传性疾病,也有散发病例。共同临床特点为肌阵挛(包括癫痫性和非癫痫性)、多种类型的癫痫发作和进行性神经功能及智能倒退。PME的肌阵挛可为多灶性、节段性或全身性,可自发出现,亦可由外部刺激或自主运动诱发,有些肌阵挛与EEG阵发性棘慢波、多棘慢波有良好的锁时相关性,提示为皮质起源的癫痫性肌阵挛;亦有些肌阵挛与EEG阵发性电活动无明显锁时相关性,推测为皮质下起源的肌阵挛。导致PME常见的疾病包括:神经元蜡样褐脂质沉积症(neuronal ceroid lipofuscinosis,NCL)、肌阵挛癫痫伴破碎红纤维病(myocolinic epilepsy with ragged red fi bers,MERRF)、唾液酸沉积症(sialidoses)、翁 -隆氏病(Unverricht-Lundborg disease)、拉福拉病(Lafora disease)、齿状核红核苍白球路易体萎缩症(dentatorubral-pallidoluysian atrophy,DRPLA)、神经型戈谢病(neuronopathic Gaucher disease)和C型尼曼-匹克病等。部分PME患者在进行多种方法的病因学检查后,仍不能明确病因。近年来随着二代测序技术在临床上的应用,新发现了多种基因变异可导致PME表型(包括GOSR2、ASAH1、KCNC1、KCTD7、TBC1D24、SCARB2、PRICKLE1、CARS2和 SERPINI1),NCL依据不同的致病基因也被分为14型(CLN1~CLN14),新基因的发现提高了对PME病因学的认识,为PME的精确诊断、预后判断及遗传咨询提供了重要依据。除成人型NCL(Parry病)、DRPLA、家族性脑病伴神经系统包涵体和KCNC1基因突变导致的PME为常染色体显性遗传以及MERRF为母系遗传外,其他PME均为常染色体隐性遗传疾病。PME的病因多为神经遗传病,尚无特效治疗方法。PME患者的肌阵挛和癫痫发作通常很难控制,多数病情呈进展性,进展情况与病因有关,多数预后不良。
(二十五)拉斯马森综合征
拉斯马森综合征(Rasmussen syndrome)又称拉斯马森脑炎(Rasmussen encephalitis),由法国医生Rasmussen等于1958年首次描述,病因和发病机制尚不清楚,可能与自身免疫因素有关,病理特征为一侧大脑半球慢性局限性炎症。主要在儿童期发病,多数在14个月~14岁发病,临床表现为局灶性运动性发作,常发展为持续性部分性癫痫发作(epilepsiapartialis continua,EPC)、进行性偏瘫和认知倒退。多数头皮EEG有癫痫样放电,但与肌肉抽动不完全同步,少数在头皮EEG没有放电。使用抽动锁定的逆向平均技术,可叠加出对侧运动区棘波或尖波,一般出现在抽动之前50毫秒以内。头颅影像学显示一侧大脑半球进行性萎缩。本病对抗癫痫发作药治疗反应差,少数对大剂量激素和丙种球蛋白治疗有短暂效果,半球手术可有效控制癫痫发作,阻止病情进展。本病预后不良,多数留有神经系统后遗症。
(二十六)热性感染相关性癫痫综合征
热性感染相关性癫痫综合征(febrile Infection-related epilepsy syndrome,FIRES)是近年来逐渐被认识的一种严重的癫痫性脑病,既往又称发热诱发的学龄儿童难治性癫痫性脑病(fever induced refractory epileptic encephalopathy in school age children,FIRES)或暴发性炎症反应癫痫综合征(fulminant inflammatory response epilepsy syndrome,FIRES)。2010年由Van Baalen首次命名定义,发病机制尚不完全清楚,近年来发现患者急性期血清白细胞介素-6(IL-6)明显升高,说明有炎症因子参与。临床特点为发病前发育正常,发病年龄2~17岁,存在前驱的热性感染,发热诱发难治性癫痫及癫痫持续状态,首次发作出现在发热后24小时~2周内(平均4~5天)。发作类型主要为局灶性或局灶继发全面性发作,发作间期意识不清,表现为嗜睡甚至昏迷。急性期经历数周或数月后,持续状态减少或停止,意识逐渐恢复进入慢性期。慢性期表现为难治性局灶性癫痫,认知减退和运动功能障碍。绝大多数患者直接从急性期到慢性期,中间缺乏静止期。脑脊液检查正常,少数伴淋巴细胞增多,蛋白正常或轻微升高,病毒及细菌学检查均阴性,没有中枢神经系统感染的直接证据。影像学缺乏特异性改变,急性期头颅MRI大多正常,仅少数可有颞叶、岛叶和基底节区异常信号。慢性期MRI多表现为脑萎缩及海马硬化,也可显示正常。脑电图急性期显示背景异常,并可见痫样放电,放电主要集中在外侧裂周围;发作时EEG提示主要累及颞叶、有时累及额叶,表现高幅慢波,部分患儿可有广泛性放电;发作间期主要表现为弥漫性慢波。缺少有效治疗方法,对多种抗癫痫发作药及免疫治疗无效,死亡率高,幸存者遗留严重的认知障碍。近年来发现白细胞介素-1(IL-1)受体拮抗剂阿那白滞素对该病治疗有效,IL-6的单克隆抗体(托珠单抗)也有一定作用,可改善患者的预后。
癫痫的病因包括先天遗传因素和后天获得性因素。随着分子遗传学、神经影像学及神经科学的快速发展,近年来癫痫病因学的研究进展很快。目前认为约30%的癫痫患者主要由明确的后天获得性因素导致,如围产期脑损伤、中枢神经系统感染、卒中、脑外伤、免疫相关的中枢神经系统疾病(免疫性脑炎、脱髓鞘疾病等)和肿瘤等。约70%的癫痫患者中遗传因素起更重要的作用。2017年ILAE提出了新的癫痫分类框架,将癫痫的病因分为六大类,包括结构性、遗传性、感染性、代谢性、免疫性和病因不明。明确癫痫的病因对治疗方案的选择和判断预后有重要意义。
一、结构性病因
结构性病因指神经影像学可见脑结构性异常,并且临床评估与影像学结合,可以推测该影像学异常很可能就是患儿癫痫发作的直接原因。结构性病因可以是获得性的,如卒中、出血、外伤、肿瘤等,也可以是遗传性的,如皮质发育畸形、结节性硬化。有些脑结构异常既可以是遗传性的,也可以是获得性的,如多小脑回畸形可能是继发于GPR56基因突变,或者获得性地继发于宫内巨细胞病毒感染。尽管这些畸形可能存在遗传性基础或由获得性病因所致,但是结构异常是患者癫痫的直接致病机制。
与结构性病因相关的综合征包括较为常见的伴海马硬化的颞叶内侧癫痫、伴下丘脑错构瘤的发笑发作、Rasmussen综合征和半侧惊厥-偏瘫-癫痫。这些结构性病因相关的综合征具有其影像学特征,也提示药物治疗多数难以控制发作,大多数需要手术治疗。
皮质发育畸形(malformation of cortical development,MCD)是癫痫和神经发育迟缓的常见原因,其种类繁多,包括局灶性皮质发育不良、多小脑回畸形、脑室周围结节状灰质异位、皮质下带状灰质异位及脑裂畸形等。这些皮质发育畸形都具有明显的遗传异质性,既可以是符合孟德尔遗传的生殖细胞单基因致病突变所致,也可以是体细胞致病性突变所致。已经确定了数十种脑发育畸形相关的基因(DCX、LIS1、DEPDC5、NPRL2、NPRL3、UBA1A、TUBB2B、TUBB3、TUBB5、TUBG1、WDR62、DYNC1H1、SLC35A2 等),这些基因突变常常干扰大脑皮质的发育。需要注意的是,结构性病因如有明确的遗传基础,如结节性硬化分别由编码错构瘤蛋白和结节蛋白的TSC1和TSC2基因突变引起,则这种癫痫为遗传性-结构性(genetic-structure)病因。
二、遗传性病因
遗传性癫痫是指癫痫由已知或推论的遗传缺陷所直接导致,并且癫痫发作是该疾病的核心症状。由此定义可以看出,确定遗传性病因(genetic etiology)主要基于两种条件之一,基于可靠的分子或细胞遗传学检测结果及分析直接诊断,或者基于既往明确的家系研究结果而推论诊断。如某患者临床表型符合Dravet综合征,通过基因检测发现SCN1A基因新发杂合致病性变异,即可以确定该患者为遗传性病因;另一种情况,如某患者临床符合儿童失神癫痫(CAE),根据既往家系研究及双生子研究的充分证据,已经公认典型CAE的病因为遗传性,因此该CAE患儿的病因可推论诊断为遗传性。遗传性病因导致的癫痫并不排除环境因素对临床表型的贡献。
癫痫的遗传性病因包括单基因遗传、多基因/复杂遗传、染色体异常及线粒体基因突变等各种遗传变异。单基因遗传是指一个基因的致病性变异就足以导致癫痫表型。符合孟德尔遗传方式,包括常染色体显性遗传、常染色体隐性遗传、X连锁遗传等。目前已知的癫痫相关致病基因与离子通道、突触形成、DNA修复、转录调控以及神经细胞内各种转运体等有关,其中离子通道相关基因最常见,主要包括编码电压门控的离子通道基因和编码配体门控的离子通道基因。随着二代测序技术的临床应用,近年来有很多癫痫综合征致病基因被发现(附录9)。2017年有学者通过PubMed、OMIM、HGMD和EpilepsyGene数据库共发现977个基因与癫痫相关,其中以癫痫为核心症状的基因有84个。截至2022年8月,已有105个发育性癫痫性脑病(DEE)相关致病基因被OMIM收录。
多基因遗传(polygenic)/复杂遗传(complex inheritance)是指多个基因的变异共同导致癫痫,每个变异都会增加癫痫的患病风险。罕见变异(特定人群中的等位基因变异频率<1%)和常见变异(特定人群中的等位基因变异频率>1%)都可能对常见遗传相关癫痫的发病以及临床表型起作用。
染色体异常是指染色体数目或结构异常,均可能导致癫痫。包括拷贝数变异、染色体异位、倒位、环形染色体等,患者常伴有发育迟缓/智力障碍,部分可伴有表观畸形。某些染色体异常以癫痫为主要表型,如环形20号染色体综合征。染色体异常区域所包含的基因是决定临床表型的重要因素。拷贝数变异(copy number variation,CNV)是人类遗传多样性的重要因素之一,约占遗传性癫痫病因的4%~10%。
目前强调任何没有找到明确获得性病因的癫痫均应考虑是否为遗传性癫痫的可能性,对于以下情况尤其需要注意:①新生儿期或婴儿期起病的癫痫(排除获得性病因);②有癫痫家族史;③病因不明的癫痫性脑病;④合并外貌异常、小头畸形、发育迟缓或孤独症表现;⑤皮质发育畸形;⑥病因不明的难治性局灶性癫痫等。
三、代谢性病因
代谢性病因是癫痫相对少见的病因,但是在婴幼儿期相对常见。代谢性癫痫的定义为已知或推测的代谢性疾病直接导致的癫痫,并且癫痫发作是该疾病的核心症状。代谢性病因是指明确的代谢缺陷伴生化改变如氨基酸代谢病、有机酸代谢病、吡哆醇依赖症、葡萄糖转运子Ⅰ缺陷等。大多数的代谢性癫痫都有遗传基础,但仍有些可能是获得性的,如脑叶酸缺乏症。许多代谢性疾病干扰脑代谢的重要功能,如能量底物的运输和利用、富含能量的磷酸盐产生、神经元和星形胶质细胞之间的代谢耦合、神经递质合成和传递和跨血脑屏障的底物运输等。还有一些代谢性疾病,积聚的代谢产物可能会直接产生神经毒性,在这类疾病中,直到有毒产物积累到足以干扰细胞功能时才会出现症状,如有机酸代谢病。其他机制包括神经元膜通透性紊乱(如全羧化酶合成酶缺乏)、底物缺乏(如丝氨酸缺乏)、金属转运障碍(Menkes病)等。
提示可能是遗传代谢病导致癫痫的线索:①新生儿或婴儿期起病的癫痫性脑病(包括婴儿痉挛症、大田原综合征以及婴儿早期肌阵挛脑病);②癫痫伴随其他神经系统症状(智力运动发育落后/倒退)或者伴全身多系统受累(肝脾大、心肌病、皮肤病变、特殊气味等);③实验室检查提示低血糖、高血氨、高乳酸或血液系统异常;脑电图提示脑病样改变(如背景慢、暴发-抑制或多灶性棘慢波);④家族史提示有同胞不明原因死亡,或者近亲结婚史。
四、感染性病因
感染性病因是指癫痫由已知的感染性事件直接导致,并且癫痫发作是疾病的核心症状。感染性病因不是指发生于急性中枢神经系统感染急性期(如脑膜炎或脑炎急性期)的症状性癫痫发作。有高达30%的中枢神经系统感染患者在疾病早期会出现癫痫发作,但这些癫痫发作在过了急性期后有可能完全缓解。癫痫的感染性病因包括脑囊虫病、结核病、人类免疫缺陷病毒(HIV)感染、脑型疟疾、亚急性硬化性全脑炎、脑弓形虫、原虫病以及先天性寨卡病毒和巨细胞病毒感染等,这些感染性病因在非洲以及南美洲的某些地区是导致癫痫的相对常见病因之一。
五、免疫性病因
免疫性病因导致的癫痫是指癫痫为自身免疫介导的中枢神经系统炎症所导致,而且癫痫发作是疾病的核心症状。近年来在儿童及成人认识到一系列有特殊表型的免疫性癫痫,急性起病的重症或者难治性颞叶癫痫以及符合自身免疫性脑炎临床综合征样表现的癫痫均应考虑做相关抗体检测。免疫性病因可以通过检测到中枢神经系统的自身免疫性炎症证据(如自身免疫抗体)或者符合具有特征性临床表现的免疫性癫痫诊断标准而确定。由于癫痫与自身免疫异常的研究不断深入,新的抗体不断被发现和可以检测,而且早期识别、早期治疗不仅能改善急性期预后,而且也能减少远期慢性癫痫的发生,因此免疫性病因越来越成为癫痫的重要病因日益受到更多的重视。
六、病因不明(unknown)
目前仍有部分癫痫患者的病因不能确定,2017年的国际癫痫分类将这些癫痫归类为病因不明的癫痫。在这一类中,只能根据基本的电临床表现,作出癫痫基本诊断。
总体来说,癫痫患者能找到病因的程度,取决于能用于病因评估资料的程度和评估手段,随着各种诊断技术的不断进步,尤其是头颅影像技术、遗传检测技术及神经免疫学的快速发展,相信越来越多的癫痫患者的病因可以被确定。明确病因才有可能进行精准治疗,因此对于所有癫痫患者,尤其是药物难治性癫痫患者,应该不断努力争取明确其病因,从而使治疗更有针对性,改善治疗效果和预后。
一、概述
从癫痫的鉴别诊断上讲,临床上的发作性事件可以分为癫痫发作和非癫痫发作。按照定义,癫痫发作的本质是脑神经元突然异常放电导致的临床表现,有一过性、反复性及刻板性的特点,脑电图显示痫样放电。癫痫发作需要与各种各样的非癫痫性发作相鉴别。非癫痫发作是指临床表现类似于癫痫发作的所有其他发作性事件。鉴别癫痫发作和非癫痫发作是癫痫诊断的首要也是最重要部分。
非癫痫发作包括心因性发作、晕厥、屏气发作、各种发作性感觉/运动/自主神经症状、睡眠障碍和感染、代谢紊乱等引起的发作性症状。非癫痫发作的原因很多,包括病理性原因和生理性原因。表2-3列出了不同年龄段常见的非癫痫性发作。
表2-3 不同年龄段常见的非癫痫性发作

| 年龄段 非癫痫性发作 |
| 新生儿和婴幼儿期 (<3岁) 呼吸异常(窒息发作/屏气发作)、运动异常(抖动或震颤/良性肌阵挛/惊跳反应/点 头痉挛/异常眼球活动)、消化系统异常(胃食管反流等) 学龄前期(3~6岁) 睡眠障碍(夜惊症/睡行症/梦魇)、交叉擦腿、惊跳反应、腹痛、注意力缺陷、晕厥 学龄期(6<~18岁) 晕厥、偏头痛及头痛、抽动症、发作性运动障碍、精神心理行为异常(焦虑/恐惧/暴 怒)、睡眠障碍 成人期(>18岁) 晕厥、癔症发作、偏头痛及头痛、舞蹈症、发作性睡病、短暂性脑缺血发作、短暂性 全面遗忘症、老年猝倒、多发性硬化发作性症状 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
二、常见非癫痫性发作与癫痫发作的鉴别
(一)晕厥(syncope)
晕厥表现为突然短暂的可逆性意识丧失伴姿势性肌张力减低或消失,由全脑血灌注量突然减少引起,并随着脑血流的恢复而正常。晕厥和癫痫发作鉴别要点见表2-4。
表2-4 晕厥和癫痫发作鉴别要点

| 鉴别要点 晕厥 癫痫发作 |
| 诱因 体位改变、持久站立、剧烈运动、情绪激动等 无诱因,或疲劳、声、光、热刺激 前驱症状 头晕、视物模糊、大汗、恶心呕吐、心悸或无明 显先兆 视觉、味觉、听觉、感觉异常等或 无前驱症状 肤色 苍白或发绀 发绀或正常 肢体情况 肢体软,偶有肢体抖动 多伴肢体强直、抽搐或无 伴尿失禁 少见 可有 发作后症状 少见 可有头痛、嗜睡 既往史 器质性心脏病或无 可有神经系统疾病或无 发作间期脑电图异常 罕见 常见 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
(二)心因性非癫痫性发作
心因性非癫痫性发作(psychogenic nonepileptic seizures,PNES)与癫痫发作鉴别要点见表2-5。
表2-5 心因性非癫痫性发作与癫痫发作的鉴别
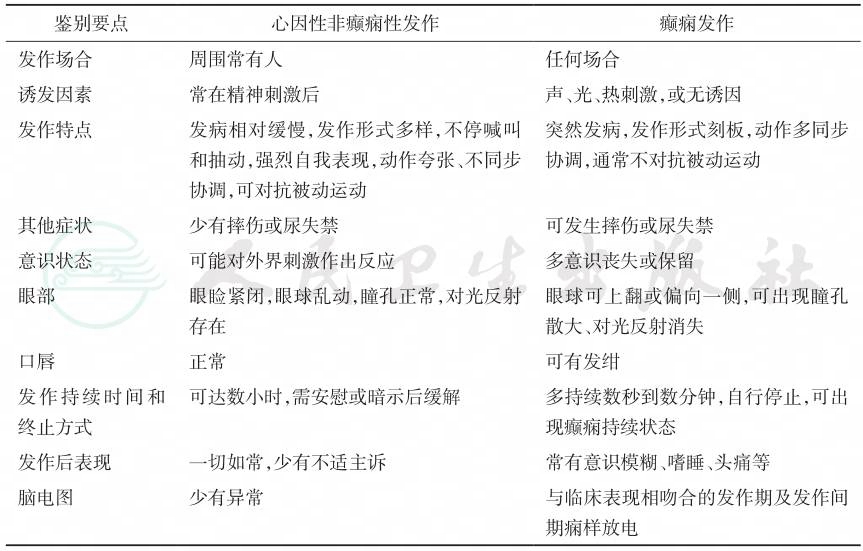
| 鉴别要点 心因性非癫痫性发作 癫痫发作 |
| 发作场合 周围常有人 任何场合 诱发因素 常在精神刺激后 声、光、热刺激,或无诱因 发作特点 发病相对缓慢,发作形式多样,不停喊叫 和抽动,强烈自我表现,动作夸张、不同步 协调,可对抗被动运动 突然发病,发作形式刻板,动作多同步 协调,通常不对抗被动运动 其他症状 少有摔伤或尿失禁 可发生摔伤或尿失禁 意识状态 可能对外界刺激作出反应 多意识丧失或保留 眼部 眼睑紧闭,眼球乱动,瞳孔正常,对光反射 存在 眼球可上翻或偏向一侧,可出现瞳孔 散大、对光反射消失 口唇 正常 可有发绀 发作持续时间和 终止方式 可达数小时,需安慰或暗示后缓解 多持续数秒到数分钟,自行停止,可出 现癫痫持续状态 发作后表现 一切如常,少有不适主诉 常有意识模糊、嗜睡、头痛等 脑电图 少有异常 与临床表现相吻合的发作期及发作间 期痫样放电 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
(三)偏头痛
癫痫和偏头痛都是发作性疾病,两者有时候需要进行鉴别。偏头痛先兆(又称偏头痛等位症)可表现为反复发作的自主神经症状,如周期性呕吐、腹型偏头痛及周期性眩晕等,伴或不伴头痛发作;而偏头痛也是癫痫常见的共患病,包括偏头痛先兆诱发的痫样发作、癫痫发作期头痛和痫性发作后头痛;因此偏头痛先兆亦需要与癫痫发作相鉴别。偏头痛与癫痫发作的鉴别要点见表2-6。
表2-6 偏头痛与癫痫发作的鉴别
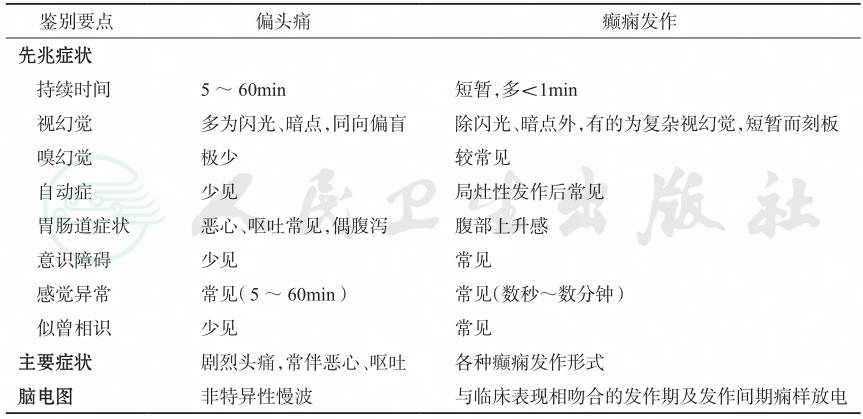
| 鉴别要点 偏头痛 癫痫发作 |
| 先兆症状 持续时间 5~60min 短暂,多<1min 视幻觉 多为闪光、暗点,同向偏盲 除闪光、暗点外,有的为复杂视幻觉,短暂而刻板 嗅幻觉 极少 较常见 自动症 少见 局灶性发作后常见 胃肠道症状 恶心、呕吐常见,偶腹泻 腹部上升感 意识障碍 少见 常见 感觉异常 常见(5~60min) 常见(数秒~数分钟) 似曾相识 少见 常见 主要症状 剧烈头痛,常伴恶心、呕吐 各种癫痫发作形式 脑电图 非特异性慢波 与临床表现相吻合的发作期及发作间期痫样放电 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
(四)屏气发作
屏气发作通常发生在6个月~6岁的婴幼儿中,高峰年龄为6~18个月。可见于正常儿童,也可见于雷特综合征(Rett syndrome)、智力障碍、遗传代谢病等患儿,这部分患儿临床症状可持续到成年期。屏气发作通常由情感伤害触发,例如疼痛、愤怒或恐惧。发作表现为短暂的啼哭,通常随后很快在用力呼气阶段发生屏气,呼吸突然停止,头后仰,躯干及肢体强直,苍白或发绀;这些症状之后常常出现瘫软和意识丧失,可能出现短暂的姿势性或强直阵挛性运动活动。儿童屏气发作的临床病程通常是良性的,大多数在8岁前停止。脑电图多数正常。哭闹也可以是癫痫发作和脑血管病卒中发作的诱因,但哭闹的剧烈程度、持续时间和呼吸停止等特点可以协助鉴别;视频脑电图监测有助于鉴别屏气发作和癫痫发作。屏气发作后心电图表现为波幅电压下降→心率变慢→变快→恢复正常,也有一定的特征性。
(五)短暂性脑缺血发作
临床多表现为神经功能的缺失性症状,如偏瘫、偏盲、偏身感觉减退等,而癫痫发作多为刺激性症状,如抽搐等。短暂性脑缺血发作多见于有脑血管病危险因素的中老年人,而癫痫在儿童和老年人均常见。
(六)睡眠障碍
睡眠障碍包括发作性睡病、睡眠呼吸暂停症、夜惊症、睡行症、梦魇、快速眼动期行为障碍、意识模糊性觉醒、节律性运动障碍、周期性睡眠增多等。而睡眠期间不愉快或不良的行为或体验亦可称为异态睡眠(parasomnia),即包括夜惊症、睡行症等。由于很多的癫痫发作类型也容易在睡眠中发病,也表现一定的运动和意识障碍等,如睡眠中发生的局灶性发作、强直-阵挛发作,某些额叶或颞叶起源的发作,且主要发生在非快速眼动期(NREM),其中夜间额叶癫痫(NFLE)需要与NREM异态睡眠相鉴别(表2-7)。
表2-7 夜间额叶癫痫(NFLE)与NREM异态睡眠鉴别诊断
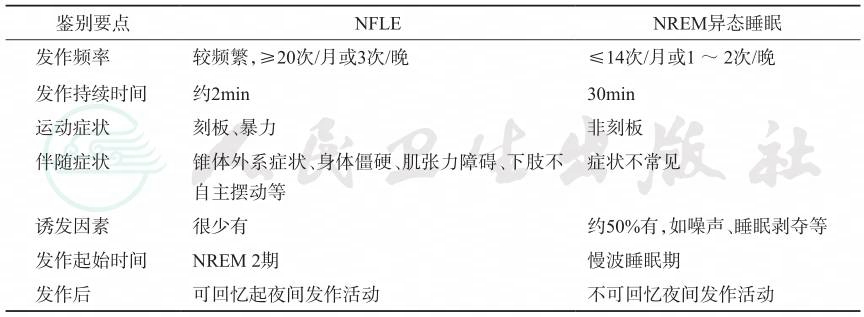
| 鉴别要点 NFLE NREM异态睡眠 |
| 发作频率 较频繁,≥20次/月或3次/晚 ≤14次/月或1~2次/晚 发作持续时间 约2min 30min 运动症状 刻板、暴力 非刻板 伴随症状 锥体外系症状、身体僵硬、肌张力障碍、下肢不 自主摆动等 症状不常见 诱发因素 很少有 约50%有,如噪声、睡眠剥夺等 发作起始时间 NREM 2期 慢波睡眠期 发作后 可回忆起夜间发作活动 不可回忆夜间发作活动 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
发作性睡病以难以控制的日间过度嗜睡、发作性猝倒、睡眠瘫痪、入睡幻觉四联症为主要临床特点。2014年国际睡眠障碍分类第3版(the third edition of the International Classif i cation of Sleep Disorders,ICSD-3)将发作性睡病分为2类:①1型,既往称为猝倒型发作性睡病,下丘脑分泌素(hypocretin)缺乏是主要原因,但少数患者有明显的下丘脑分泌素的降低而没有猝倒的表现。突出的病理生理特征是中枢神经系统食欲素(下丘脑分泌素-1)不足,这是维持警觉所必需的一种肽。②2型,患者有嗜睡表现,并且可能存在入睡前幻觉和睡眠瘫痪,但不伴猝倒,且测定脑脊液中下丘脑分泌素水平无显著下降。发作性睡病的猝倒发作有时易与失张力或肌阵挛发作混淆,但一般发作性睡病可追忆猝倒发作,视频-睡眠多导监测是鉴别睡眠障碍和癫痫发作最可靠的方法。
(七)抽动障碍
抽动障碍需要和癫痫发作(如肌阵挛)相鉴别。鉴别要点见表2-8。
表2-8 抽动障碍和癫痫肌阵挛发作的鉴别

| 鉴别要点 抽动障碍 癫痫肌阵挛发作 |
| 发病年龄 5~10岁多见 任何年龄 临床特征 一组或多组肌肉突发、重复和刻板性不随意抽 动,通常是非节律性,多见于面、颈、肩及上肢 反复节律性快速抽动,可涉及多 组肌肉,呈同步性 受意识控制 可能短时有效 无效 睡眠 症状减轻或消失 基本无影响 发作时意识状态 清楚 清楚、迟钝或丧失 脑电图 正常或与抽动无关的背景慢波 慢波或痫样放电 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
一、区分诱发性和非诱发性癫痫发作
并非所有的癫痫发作都要诊断为癫痫。按照定义,患者的发作必须是非诱发性癫痫发作时才能诊断癫痫,而诱发性癫痫发作即使反复出现通常也不考虑诊断为癫痫。把反复的急性症状性发作误诊为“症状性癫痫”的做法必然导致过度诊断及治疗,也会导致癫痫流行病学调查结果不可靠。有癫痫发作但通常不诊断为癫痫的情况包括:新生儿良性发作、热性惊厥、酒精或药物戒断性发作、中枢神经系统或全身系统性疾病的急性期出现的发作,如自身免疫性脑炎的急性症状性癫痫发作。按照2014年ILAE癫痫临床实用性定义(表2-9),反复发生的反射性发作(通过视觉、听觉、躯体感觉或躯体运动刺激,或通过更高的皮质功能活动诱发)可以诊断为反射性癫痫,尽管每次发作看似是“诱发性”的。
表2-9 癫痫的临床实用性定义(ILAE,2014)

| 癫痫是一种脑部疾病,符合如下任何一种情况可确定为癫痫: |
| 1. 至少两次间隔>24h的非诱发性(或反射性)发作 2. 一次非诱发性(或反射性)发作,并且在未来10年内,再次发作风险与两次非诱发性发作后的再发风险 相当时(至少60%) 3. 诊断为某种癫痫综合征 |
| 符合如下任何一种情况,可认为癫痫诊断可以解除: |
| 1. 已经超过了某种年龄自限性癫痫综合征的患病年龄 2. 已经10年无发作,并且近5年已停用抗癫痫发作药 |
引自:临床诊疗指南——癫痫病分册(2023修订版).第1版.ISBN:978-7-117-34579-8
二、病史和辅助检查在癫痫诊断中的作用
病史资料是诊断癫痫最重要的依据,癫痫在很大程度上是一种临床诊断。按照定义,临床出现两次非诱发性癫痫发作时就可以诊断癫痫了,通常也就可以考虑药物治疗了。多数情况下,详细询问病史尤其是发作史就可确定发作性症状是否为癫痫性发作,甚至可以初步进行发作类型和癫痫(综合征)类型的诊断,后期的脑电图及影像学检查往往作为进一步验证或明确前期诊断的手段。脑电图异常不一定要诊断癫痫,脑电图正常也不能排除癫痫。应避免患者短期内已有数次典型的大发作,但因脑电图正常而未能诊断癫痫并延误治疗的情况。
三、获取完整的癫痫发作史
病史采集不充分是造成癫痫误诊的最常见原因。癫痫发作往往历时短暂,医师目睹癫痫发作的可能性不大,所以详细而有条理的病史询问尤为重要。应建议患者本人和发作目击者一同就诊,以便获取完整的病史。当患者就诊时描述不清楚,医师电话询问发作目击者很有必要。如有可能,建议患者或家属用手机或家用摄像机把发作过程摄录下来,就诊时供医师分析。另外,在患者或目击者表述不清的情况下,让他们观看各种典型的发作录像也是很好的方法,往往可以使他们找到与患者表现最类似的发作。如果确实难以获得可靠病史,应向患者解释病史的重要性,以便再次发作时留意观察及复诊时提供。
四、避免漏掉“轻微发作(minor seizures)”
完整的发作类型信息对于癫痫(综合征)的类型诊断很重要。在询问病史时,既要关注表现明显的发作(如全面性强直-阵挛发作),也要关注患者或发作目击者经常忽略或不主动告之的某些“轻微发作”,例如,先兆发作、肌阵挛发作、意识障碍轻微的局灶性发作等。举例:对于主诉有过数次全面性强直-阵挛发作且既往史正常的青少年患者,如果病史中能够问出常被患者忽视的晨起后肢体“抖动”的情况,则临床要考虑“青少年肌阵挛癫痫”的可能,否则考虑可能是“仅有全面强直-阵挛发作的特发全面性癫痫”。
五、长程视频脑电图监测的应用
按照定义,诊断癫痫发作的“金标准”应该依据发作期异常脑电活动和临床表现的时间相关性,这可通过长程视频脑电图监测来实现。当然,所有患者都进行长程监测既不实际也无必要。对于通过详细病史询问仍不明确发作性质的病例,可以进行长程视频脑电图监测来明确诊断。另外,对于目前国内容易诊断诸如“腹型癫痫”“头痛型癫痫”“以××为唯一发作表现的癫痫”现象,也应以上述的“金标准”来衡量和检验。当然,实践中也应了解长程视频脑电图监测的局限性和不足。
六、识别“假性”药物难治性癫痫
在诊断药物难治性癫痫之前,应注意排除是否为“假性”药物难治性癫痫。重点考虑有无如下可能:①非癫痫性发作;②癫痫发作的分类错误(如将失神发作误诊为局灶性发作);③针对发作类型的选药不当(如用卡马西平控制失神发作);④药物剂量不足或给药方法不当;⑤患者服药依从性差;⑥加重发作的可控诱因(如过量饮酒、缺少睡眠等);⑦其他可导致癫痫难治的病因(如维生素B6依赖症、葡萄糖转运体Ⅰ缺陷症等)。另外,有些癫痫患者可能同时存在癫痫发作和非癫痫发作,应注意鉴别,必要时行长程视频脑电图监测明确诊断。避免因为将发作性症状都误认为是癫痫发作,而不断增加药物剂量或频繁更换药物来控制“难治性癫痫”的情况。
七、癫痫诊断和治疗间的关系
癫痫的诊断和治疗尽管关系密切,但不一定存在必然联系。一方面,诊断癫痫后不一定都要治疗。例如,对发作稀疏的儿童良性局灶性癫痫或发作轻微(如仅有先兆发作)癫痫患者,可以选择不治疗。对癫痫患者是否治疗取决于多方面因素,包括患者意愿、个体化服药/不服药的获益-风险比等。另一方面,不诊断癫痫也可考虑开始治疗。例如,对于脑炎急性期出现的反复癫痫发作患者,尽管不诊断为癫痫,临床上通常会进行药物治疗。
影响癫痫的预后因素包括癫痫的自然病史、病因、病情和治疗情况等。由于大多数癫痫患者(尤其在发达国家)在诊断后接受了治疗,有关癫痫自然病程的认识还很少。总体看来,大多数癫痫患者抗癫痫发作药治疗的预后较好,约2/3病例可获得长期的发作缓解,其中部分患者可完全停药仍长期无发作。
一、新诊断的癫痫预后
1.经治疗的新诊断的癫痫预后
通常情况下,在出现两次及以上非诱发性癫痫发作时才诊断癫痫,并开始药物治疗。在随诊观察10年和20年时,经治疗的癫痫累积5年发作缓解率分别为58%~65%和70%。在随诊50年时,有97.1%患者经历了至少1年的无发作期,2年、5年和10年无发作缓解率依次为89.5%、77.1%和44.4%。在随诊10年时,经治疗的成人癫痫5年发作缓解率为61%。在随诊12~30年时,经治疗的儿童癫痫3~5年发作缓解率为74%~78%。对于儿童期发病的癫痫患者,在随诊30年时,有64%的病例可以达到5年终点无发作,其中74%的患者停用了药物。
2.新诊断的癫痫预后的主要影响因素
最主要的影响因素是癫痫的病因。总体上,癫痫早期的发作频率少、全面性强直-阵挛发作、无精神共患病者更容易达到发作缓解。在儿童癫痫中,能找到明确癫痫病因、首次发作年龄小的患者预后相对较差。其他影响癫痫预后的因素有脑电图是否有局灶性慢波或癫痫样放电、首次发作后6个月内出现再次发作的次数等。一般认为,性别对预后影响不大。
3.癫痫综合征的预后
根据综合征的本身性质和对治疗的反应,癫痫综合征的预后大体上可分为如下四种:
(1)预后很好:
约占20%~30%,属良性癫痫。通常发作稀疏,可以自发缓解,不一定需要药物治疗。这类综合征包括新生儿良性发作、自限性局灶性癫痫(儿童良性癫痫伴中央颞区棘波/儿童良性枕叶癫痫等)、婴儿良性肌阵挛癫痫以及某些有特殊原因促发的癫痫。
(2)预后较好:
约占30%~40%。癫痫发作很容易用药控制,癫痫也有自发缓解的可能性。这类综合征包括儿童失神癫痫、仅有全面强直-阵挛性发作的癫痫和某些局灶性癫痫等。
(3)药物依赖性预后:
约占10%~20%。抗癫痫发作药能控制发作,但停药后容易复发。这类综合征包括青少年肌阵挛癫痫、大多数局灶性癫痫(结构性或病因不明)。
(4)不良预后:
约占20%。尽管进行了积极的药物治疗,仍有明显的癫痫发作,甚至出现进行性神经精神功能衰退。这类综合征包括各种癫痫性脑病、进行性肌阵挛癫痫和某些症状性局灶性癫痫。
4.抗癫痫发作药治疗和癫痫发作的预后
目前的证据显示,抗癫痫发作药治疗通常只能控制发作,不能阻止潜在致痫性(epileptogenesis)的形成和进展。一线抗癫痫发作药之间没有明显的疗效差别。如果正确选择抗癫痫发作药,新诊断癫痫患者的无发作率能达到60%~70%。有研究显示,使用第一种单药治疗后有49.5%的新诊断癫痫患者能达到无发作,再使用第二种及第三种单药治疗时则仅有13.3%和3.7%的患者可达到无发作。如果单药治疗效果不佳,可考虑联合用药。但即使经过积极治疗,新诊断的癫痫患者中有约20%~30%发作最终控制不佳。
二、停药后癫痫的预后
1.停药后癫痫的复发情况
在减药过程中或停药后,癫痫复发的风险从12%~66%不等。既往荟萃分析显示,停药后1年和2年的复发风险分别为25%和29%。在停药后1年和2年时,保持无发作的患者累积比例在儿童中分别是66%~96%和61%~91%,而在成人中则分别是39%~74%和35%~57%,说明成人癫痫要比儿童癫痫的复发率高。复发比例在停药后12个月内最高(尤其是前6个月),随后逐渐下降。近期荟萃分析表明,停药后5.3年(IQR 3.0~10.0年)癫痫的复发率为46%。有报道停药后复发的患者中,约10%~20%再次启动抗发作治疗后不能达到发作完全缓解。
2.停药后癫痫复发的预测因素
停药后癫痫复发的预测因素包括:发作完全缓解前癫痫病程较长、停药前发作完全控制时间较短、有热性惊厥史、发作完全缓解前的发作次数较多、非自限性癫痫综合征、发育落后及停药前EEG可见痫样异常。
停药后远期无发作(无论是否复发)的预测因素包括:发作完全缓解前癫痫病程较短、停药前发作完全控制时间较长、停药前使用抗癫痫发作药的种类数较少、男性患者、无癫痫家族史、发作完全缓解前的发作次数较少、无局灶性发作、停药前EEG无异常。
值得注意的是,延缓停药时间(增加停药前无发作年数)可降低复发风险。
[1]中国抗癫痫协会.临床诊疗指南:癫痫病分册[M].北京:人民卫生出版社,2015.
[2]FISHER R,ACEVEDO C,ARZIMANOGLOU A,et al.A practical clinical definition of epilepsy[J].Epilepsia,2014,55(4):475-482.
[3]BLUME W T,LUDERS H O,MIZRAHI E,et al.Glossary of descriptive terminology for ictal semiology:report of the ILAE task force on classif i cation and terminology[J].Epilepsia,2001,42(9):1212-1218.
[4]FISHER R S,CROSS J H,FRENCH J A,et al.Operational classification of seizure types by the International League Against Epilepsy:Position Paper of the ILAE Commission for Classification and Terminology[J].Epilepsia,2017,58(4):522-530.
[5]PRESSLER R M,CILIO M R,MIZRAHI E M,et al.The ILAE classif i cation of seizures and the epilepsies:Modif i cation for seizures in the neonate.Position paper by the ILAE Task Force on Neonatal Seizures[J].Epilepsia,2021,62(3):615-628.
[6]FISHER R S,BOAS W V,BLUME W,et al.Epileptic seizures and epilepsy:def i nitions proposed by the International League against Epilepsy(ILAE)and the International Bureau for Epilepsy(IBE)[J].Epilepsia,2005,46(4):470-472.
[7]LUDERS H,ACHARYA J,BAUMGARTNER C,et al.Semiological seizure classif i cation[J].Epilepsia,1998,39(9):1006-1013.
[8]Proposal for revised classif i cation of epilepsies and epileptic syndromes.Commission on Classif i cation and Terminology of the International League Against Epilepsy[J].Epilepsia,1989,30(4):389-399.
[9]BERG A T,BERKOVIC S F,BRODIE M J,et al.Revised terminology and concepts for organization of seizures and epilepsies:report of the ILAE Commission on Classif i cation and Terminology,2005–2009[J].Epilepsia,2010,51(4):676-685.
[10]SCHEFFER I E,BERKOVIC S,CAPOVILLA G,et al.ILAE classification of the epilepsies:Position paper of the ILAE Commission for Classif i cation and Terminology[J].Epilepsia,2017,58(4):512-521.
[11]WANG J,LIN Z J,LIU L,et al.Epilepsy-associated genes[J].Seizure,2017,44:11-20.
[12]BAYAT A,BAYAT M,RUBBOLI G,et al.Epilepsy syndromes in the fi rst year of life and usefulness of genetic testing for precision therapy[J].Genes(Basel),2021,12(7):1051
[13]TSUCHIDA N,NAKASHIMA M,KATO M,et al.Detection of copy number variations in epilepsy using exome data[J].Clinical genetics,2018,93(3):577-587.
[14]SHARMA S,PRASAD A N.Inborn Errors of Metabolism and Epilepsy:Current Understanding,Diagnosis,and Treatment Approaches[J].Int J Mol Sci,2017,18(7):1384.
[15]VEZZANI A,FUJINAMI R S,WHITE H S,et al.Infections,inflammation and epilepsy[J].Acta neuropathologica,2016,131(2):211-234.
[16]SULEIMAN J,DALE R.The recognition and treatment of autoimmune epilepsy in children[J].Dev Med Child Neurol,2015,57(5):431-440.
[17]BEGHI E,GIUSSANI G,SANDER J W.The natural history and prognosis of epilepsy[J].Epileptic Disord,2015,17(3):243-253.
[18]BEGHI E,BERETTA S,CARONE D,et al.Prognostic patterns and predictors in epilepsy:a multicentre study (PRO-LONG)[J].J Neurol Neurosurg Psychiatry,2019,90(11):1276-1285.
[19]BRORSON L O,ERIKSSON M,BLOMBERG K,et al.Fifty years’ follow-up of childhood epilepsy:Medical outcome,morbidity,and medication [J].Epilepsia,2019,60(3):381-392.
[20]BRODIE M J,BARRY S J,BAMAGOUS G A,et al.Patterns of treatment response in newly diagnosed epilepsy[J].Neurology,2012,78(20):1548-1554.
[21]LAMBERINK H J,OTTE W M,GEERTS A T,et al.Individualised prediction model of seizure recurrence and long-term outcomes after withdrawal of antiepileptic drugs in seizure-free patients:a systematic review and individual participant data meta-analysis [J].The Lancet Neurology,2017,16(7):523-531.
[22]WIRRELL E C,NABBOUT R,SCHEFFER I E,et al.Methodology for classification and definition of epilepsy syndromes with list of syndromes:Report of the ILAE Task Force on Nosology and Def i nitions [J].Epilepsia,2022,63(6):1333-1348.
北京市朝阳区潘家园南里19号人卫大厦
© 2021 人民卫生出版集团 北京人卫智数科技有限公司 京ICP备:19054460号 京公网安备 11010502039674号
京公网安备 11010502039674号

下载APP

公众号

营业执照

出版物经营许可证

 收藏
收藏 已收藏
已收藏



