


 收藏
收藏 已收藏
已收藏英文名称 :idiopathic pulmonary fibrosis
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是原因不明的慢性进行性致纤维化间质性肺炎,主要见于中老年男性,局限于肺,临床上呈渐进性呼吸困难和肺功能恶化,组织病理学和胸部HRCT表现为特征性的寻常型间质性肺炎(UIP)。
近年来关于IPF发病机制研究,最重要的进展是从“慢性炎症驱动说”转向“肺泡上皮细胞微损伤说”(图1)。易感者和老年人肺泡上皮细胞(AECs)因反复微损伤发生凋亡,引发血管外凝血、免疫系统、成纤维细胞和其他细胞激活等一系列创伤-修复反应,而ACEs在原始刺激消失后仍持续激活,局部成纤维细胞迁移和增殖,循环成纤维细胞朝向损伤部位聚集和分化,形成UIP组织学上特征性标志的“肌成纤维细胞灶”,并分泌大量细胞外基质(ECM),后者沉积到间质和肺泡腔,致使肺结构进行性破坏和功能丧失。此种病理级联反应涉及细胞与细胞、细胞与基质之间相互作用,有许多生化介质的参与和调节,其中转化生长因子β(TGF-β)参与多个环节与过程,在IPF形成中是关键性细胞因子。
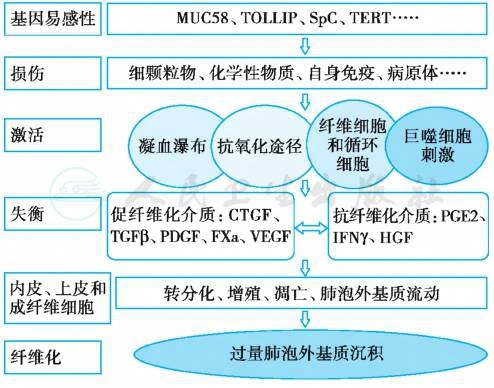
图1 特发性肺纤维化发病机制模式
注:MUC58.黏蛋白基因;TOLLIP.TOLL样受体基因;SpC.表面活性蛋白 C基因;TERT.端粒酶基因;CTGF.结缔组织生长因子;FⅩa.Ⅹa因子;HGF.肝细胞生长因子;IFN-γ.γ-干扰素;PDGF.血小板生长因子;PGE2.前列腺素 E2;TGF-β.转化生长因子 β;Th.辅助性T细胞;VEGF.血管内皮生长因子。
致ACEs损伤的危险因素包括吸烟、病毒感染、空气污染、职业暴露、微吸入、老年化等。IPF易感性与某些基因变异/突变和转录改变有关,已经发现IPF风险基因变异/突变10余种,主要黏蛋白基因MUC5B、肺表面活性蛋白C和A(SPC和SPA)基因、端粒酶基因(TERT、TERC)等。
IPF在全球均有发生。患病率和年发病率分别为(0.5~27.9)/10万和(0.22~8.8)/10万。中国大陆尚无IPF的流行病学数据。1999—2009年间怀疑间质性肺疾病(ILD)接受外科肺活检共 418例,病理诊断 IPF有 61例,占总病例数的14.8%和病理诊断ILD的35.4%。中国台湾地区1997—2007年根据医保记录回顾性研究提示,IPF年发病率为(0.9~1.6)/10万,患病率自2000—2007年增加超过2倍,中位生存期按广义和狭义(有CT和活检)定义分别为0.9年和0.7年,死因序位仅次于癌症。
IPF的病理改变表现为UIP。IPF炎症轻微,若有大量炎症细胞聚集,则需要考虑其他诊断。UIP虽然是IPF的病理特征,但两者不是同义词,风湿病肺、石棉沉着病和药物性肺病后期病理改变亦呈现UIP。
(一)血液检查
晚期患者血液红细胞和血细胞比容增加。多数患者血沉增高,10%~20%患者循环抗核抗体(ANA)和类风湿因子(RF)低滴度阳性。
(二)高分辨率CT(HRCT)
是诊断IPF的必备检查。技术上要求尽快速度扫描以减少呼吸运动对图像的影响,于深吸气末和深呼气仰卧位各采集1次图像,薄层重建(≤1.5mm)。根据HRCT表现分为4型(表1),以界定IPF的影像学诊断术语及其标准。
表1 UIP的HRCT诊断标准
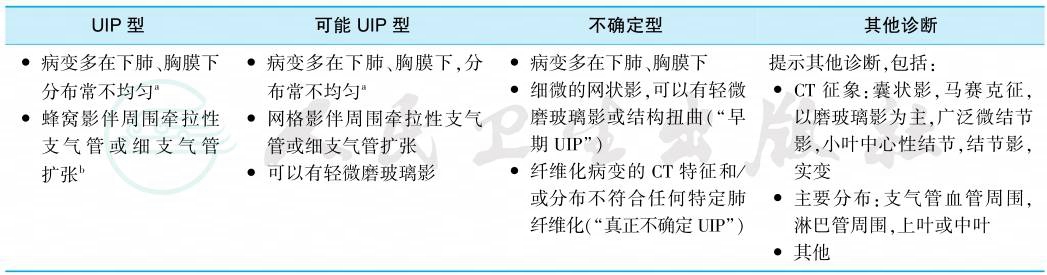
注:a病变空间分布存在多样,偶呈弥漫性分布,亦可两肺病变不对称;b合并其他CT表现:轻微磨玻璃影,网格影及肺骨化。
(三)肺功能测定
IPF的特征性肺功能改变是肺容量减少,用力肺活量(FVC)降低,弥散量(DLCO)降低,肺泡-动脉氧分压差(PA-aO2)增宽,肺顺应性降低,心肺运动试验异常。弥散量降低和PA-aO2增宽是 IPF的早期异常。定期、连续用力肺活量(forced vital capacity,FVC)监测,其下降趋势对预测死亡优于其他肺功能参数。FVC下降10%或DLCO下降15%为有意义;在6分钟步行试验氧饱和度低于88%和IPF合并肺气肿的患者连续DLCO监测较FVC更敏感;6分钟步行试验对预测肺动脉高压和肺移植时间有参考价值。
(四)外科活检、支气管镜检查和组织病理学诊断
与过去主张不同,现在仅在HRCT上UIP不确定时才推荐外科肺活检,或考虑BAL和TBLB。
依据组织病理学所见,并保持HRCT分型一致,分为4型:①UIP型:显示致密纤维化伴肺结构扭曲,纤维化主要位于胸膜下或间隔旁,肺实质受纤维化累及而呈斑片状,成纤维细胞灶,无其他疾病的病理特征;②可能UIP型:具有UIP型的某些特征,但不满足UIP/IPF确诊的全部特征;③不确定型:纤维化伴或不伴结构扭曲,其特征更符合其他非UIP间质病或继发于其他原因的UIP,或者虽然具有UIF型的某些特征,但有提示“其他诊断”的特征;④其他诊断:可见其他肺间质病的特征(如没有肌成纤维细胞灶或疏松纤维化),或具有其他疾病的组织学特征。
(一)药物治疗
1.N-乙酰半胱氨酸(N-acetylcystenine,NAC)
NAC 作为抗氧化剂治疗IPF有争议,但有荟萃分析显示NAC治疗减缓FVC和DLCO的下降,疾病进展趋缓。NAC可以雾化吸入给药,还可以与抗纤维化药物联合使用。曾经广泛使用NAC、泼尼松和硫唑嘌呤三联治疗IPF,已证明该方案无益,甚至有害。糖皮质激素亦被确定无效,但在急性加重和某些难以控制的症状如顽固性咳嗽,激素仍是经验性治疗的可选药物。
2.吡非尼酮(pirfenidone)
吡非尼酮抑制 TGF-β生成及其下游信号,抑制胶原合成和成纤维增殖。吡非尼酮治疗IPF的疗效体现在FVC下降减缓,全因死亡率和IPF相关死亡率降低。长期服用吡非尼酮患者的问卷调查显示,1年内停药数达37.4%,停药原因以不良反应最常见(41.4%),其次是死亡(32.3%)。治疗2年患者FVC改善率较小,但无恶化,提示吡非尼酮治疗可以维持FVC长期稳定。不良反应多为消化道症状、肝功能损害、皮疹和光敏反应等,发生率及其严重程度报道不一。国产吡非尼酮为胶囊,每粒100mg,推荐用法为成人初始剂量200mg/次、3次/d,餐后服用,希望能在2周内通过每次增加200mg,最终维持在每次600mg/次、3次/d。美国和欧盟吡非尼酮制剂为267mg/胶囊,推荐用法为第1~7天1粒/次、3次/d(801mg/d),第 8~14 天 2 粒/次、3 次/d(1 602mg/d),第15天以后 3粒/次、3次/d(2 403mg/d)。
3.尼达尼布(nintedanib)
本品是酪氨酸激酶抑制剂,可抑制血管内皮生长因子、成纤维细胞生长因子和血小板源生长因子受体,抑制成纤维细胞向肌成纤维细胞的增殖、迁移和转化,并有一定的抗炎作用。临床试验证明,其可延缓IPF患者的FVC下降。尼达尼布减少IPF急性加重的频率,但不降低急性加重的死亡风险。疗效不依赖于年龄、性别、吸烟状况和FVC基线值;白种人和亚洲人的疗效相似。尼达尼布持续治疗可以稳定患者的肺功能直至肺移植,而且不增加术后并发症和病死率。尼布尼达最常见不良反应是腹泻,其他有恶心、呕吐、体重减轻、转氨酶升高等,皮肤不良反应则明显少于吡非尼酮。应用时应警惕出血和血栓形成的风险,避免使用抗凝剂或促血栓形成的药物。推荐减量至150mg/次、2次/d。若有腹泻等不良反应,可减为100mg/次、2次/d。
吡非尼酮和尼布尼达是目前治疗IPF仅有的两个被证明可以获益(延缓肺功能下降)的药物,许多具体实践问题仍待更多研究。倾向于IPF一旦诊断即可用药,早期用药可能更加获益;两药疗效相近,药物选择取决于临床医师经验、药物不良反应和患者可耐受性的评估;疗程无统一规定,长期治疗可以保持持续有效;两药联合理论上有协同作用,目前缺少临床证据,而且可能增加不良反应;当一种药物疗效不佳时,更换另一药物仍可以有效。
4.抗酸剂
质子泵抑制剂或组胺拮抗剂适用于合并胃食管反流病的患者。
(二)非药物治疗
1.肺移植
肺纤维化肺移植的指征是:①组织病理学诊断UIP或非特异性间质性肺炎(NSIP);②FVC<80%预计值或DLCO<40%预计值;③气急和肺功能受限确定由肺部病疾病所致;④需要氧疗,即使仅在活动时需要;⑤其他治疗无效。
等候肺移植的指征是:①6个月随访肺功能 FVC下降>10%;②6个月随访DLCO下降>15%;③6个月随访6分钟步行试验显示氧饱和度<88%、距离<250m或减少>50m;④肺动脉高压;⑤由于病情恶化、气胸或急性加重住院。
肺移植可以使IPF患者生存期延长,部分患者生活质量改善。目前IPF患者肺移植术后5年生存率约50%,而IPF患者诊断后的3~5年生存率仅为30%~35%。年轻特别是肺动脉压较高的患者可以优先考虑双肺移植。
2.长期氧疗
根据长期氧疗在其他慢性低氧血症肺部疾病患者的治疗获益,IPF国际指南推荐在静息状态下存在临床明显缺氧的患者应用长期氧疗。
3.肺康复
在包括IPF在内慢性致残性肺部疾病治疗中,肺康复内容主要是通过训练提高运动耐力、改善营养状况、心理社会支持和患者教育。肺康复改善IPF患者运动耐力、症状及其对活动能力的影响,使健康相关生活质量评分增加。









