


 收藏
收藏 已收藏
已收藏患儿,男,4岁5个月。
主诉:间断发热20余天。
现病史:20余天前患儿出现发热,当时体温39℃,不伴手脚凉,无寒战,无抽搐,无咳嗽,无喘息,无周身皮疹,予患儿口服美林后,热可将至正常,每4~6小时发热1次,家长予患儿口服头孢(具体不详)、维C银翘片、清开灵2天后患儿发热未见好转,且口服退热药物后热退不明显,就诊于当地医院,静脉滴注氨曲南8天,静脉输液期间间断予地塞米松退热,停输液2天后患儿再次出现发热,10天前发热时出现寒战,体温最高40.8℃,无抽搐,伴声咳,少许痰,无喘息,无流通及咽痛。于家中间断口服美林退热,6~7小时体温复升,同时口服头孢克肟、清开灵、抗病毒口服液,于急诊科应用地塞米松退热后收入笔者科室。患儿病来精神状态可,发热时精神状态稍差,病程中无皮疹,食欲不佳,大、小便正常。
既往史:既往体质差,患儿2岁以后每年“感冒”约7~8次,均有发热表现,多次患急性中耳炎及鼻窦炎,且大多需要静脉滴注抗生素治疗。2.5岁时因化脓性扁桃体炎住院15天后治愈出院;3.5岁时患麻疹,住院10余天后好转出院;本次入院前1个月因急性阑尾炎入住笔者医院儿普外科,行阑尾切除术,过程顺利。
过敏及接触史:无食物及药物过敏史。无肝炎、结核等传染病接触史。
个人及家族史:患儿系G1P1,足月,剖宫产,出生无抢救窒息史,生长发育同同龄儿,疫苗接种不全(具体不详)。否认家族遗传代谢性疾病史。
入院查体及相关检查:体温37.2℃;脉搏126次/min;呼吸28次/min;血压100/65mmHg。神志清楚,状态反应可,面色及甲床苍白,周身皮肤黏膜未见黄染及出血点。颈部可触及浅表淋巴结肿大,蚕豆大小,活动可,无压痛。呼吸平稳,口唇红润。咽部充血,双扁桃体Ⅰ度肿大,气管居中。双肺听诊呼吸音粗,双肺未闻及明显干、湿啰音。心音有力、律齐,各瓣膜听诊区未闻及病理性杂音。腹部软,右下腹5cm术后瘢痕,腹不胀,无胃肠型及蠕动波,无触痛,肝脾肋下未触及。四肢活动可,脉搏有力,神经系统查体未见异常,四肢末梢温,指甲移行处无脱皮,CRT 3秒。
辅助检查:门诊急检血常规:WBC 5.5×109/L,NE%8.7,N 0.48×109/L;RBC 4.5×1012/L,HGB 101g/L,HCT 30.9%,MCV 69.8fl,MCH 21.9pg,MCHC 315g/L,PLT 446×109/L。CRP 121mg/L。
入院后因诊断尚不明确,根据病情分析完善相关检查,同时给予对症支持治疗:①注意休息,加强营养;②及时退热,适当补液。入院后患儿仍有发热,予患儿头孢呋辛和阿奇霉素控制感染。
检查及结果分析:
1.入院后完善各项检查 血常规:WBC 2.0×109/L,NE%22.2,N 0.44×109/L,RBC 4.43×1012/L,HGB 96g/L,HCT 30.7%,MCV 69.3fl,MCH 21.7pg,MCHC 313g/L,PLT 384×109/L,未见异型淋巴细胞及幼稚细胞,仍提示粒细胞缺乏及轻度小细胞低色素性贫血;尿常规正常;便常规正常,潜血阴性;肝肾功能正常,血清胆红素正常。血气分析正常。
2.血CRP 122mg/L(0~8mg/L);PCT 0.215ng/L(< 0.05ng/L);ESR 40mm/h,均增高。ASO<25.0(0~200U/ml),正常;病原学检测显示肺炎支原体抗体阴性,肺炎支原体抗体-IgM(MPAbIgM)阴性、鼻咽拭子肺炎支原体DNA测定阴性,肺炎衣原体抗体-IgM、结核抗体(TBAb)、常见呼吸道、肠道病毒及肝炎病毒检测均阴性,血细菌培养未见细菌生长,结核菌素试验阴性,提示除既往有肺炎支原体感染外,无结核等其他感染迹象。
3.铁蛋白375.7ng/ml(11~336.2ng/ml),增高;骨穿(图1)显示增生明显活跃骨髓象,粒红比例倒置,组织细胞比值增高。
4.免疫球蛋白,IgG 0.2g/L(正常4.81~12.21g/L),IgA 0.3g/L(正常0.42~1.58g/L),IgM 1.9g/L(正常 0.41~1.65g/L)。淋巴细胞亚群:总T细胞(%)68(55~84),T抑制毒细胞(%)45(13~41),T辅助细胞(%)12(31~60),Th/Ts 0.27(0.71~2.78),NK细胞(%)16(7~36),总B细胞(%)13(5~20)均正常,提示免疫缺陷病可能。总IgE<17.1U/ml(0~90U/ml)正常。补体C3 1.9g/L(0.9~1.8g/L)略高,补体 C4 0.2g/L(0.1~0.4g/L)正常。
5.RF、抗心磷脂抗体(ACA)、抗中性粒细胞胞质抗体测定(ANCA)及抗核抗体系列(ANA)均阴性,初步除外结缔组织病引起患儿发热的可能。
6.胸部CT示(图2)右肺上叶尖段及左肺下叶基底段少许炎症。腹部CT未提示腹腔占位及积液表现。心电图正常,心脏、肝胆脾及肾脏彩超未发现异常。脑电图正常。
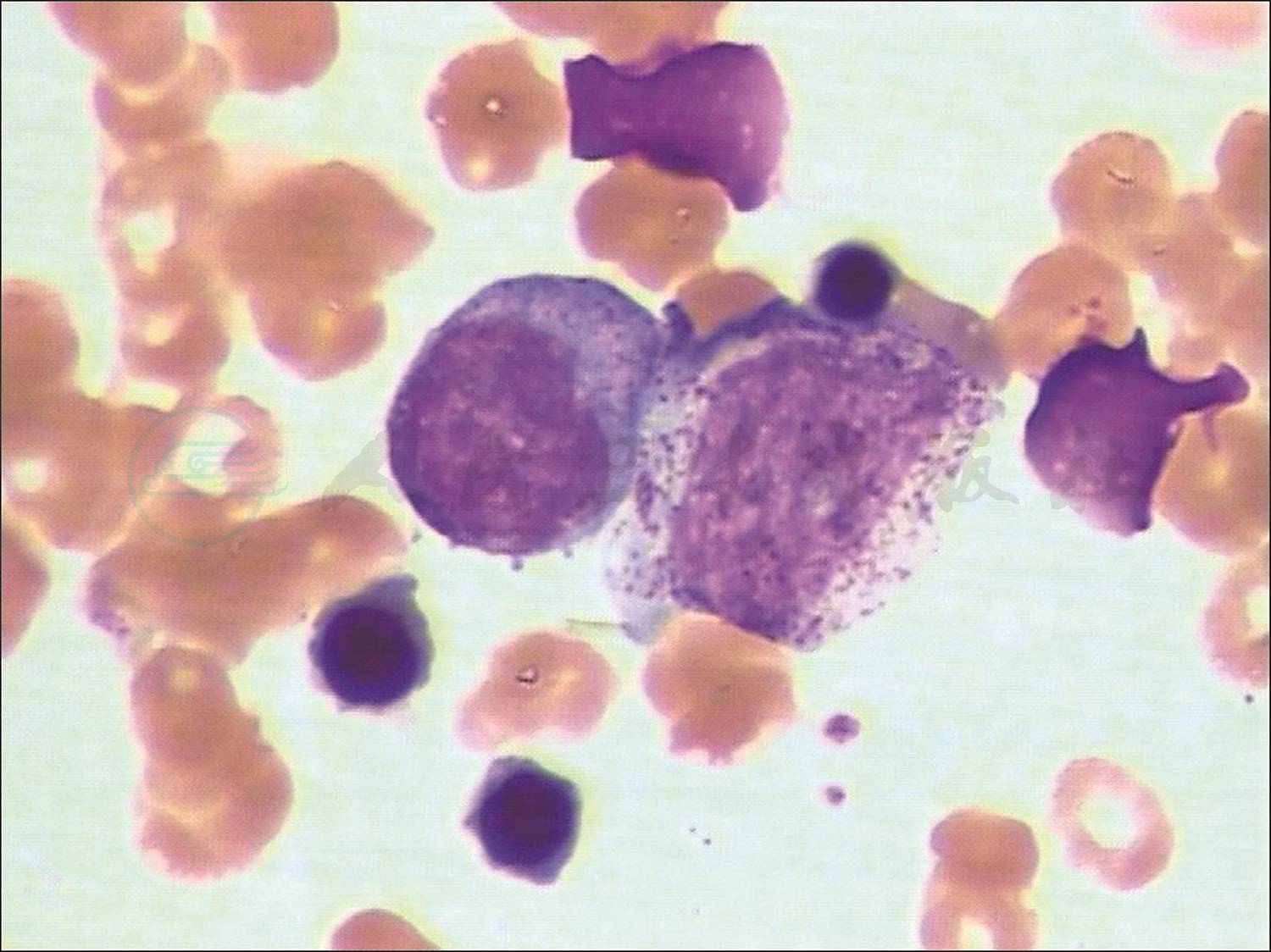
图1 骨髓穿刺术
结果显示:增生明显活跃骨髓象,粒红比例倒置,组织细胞比值增高
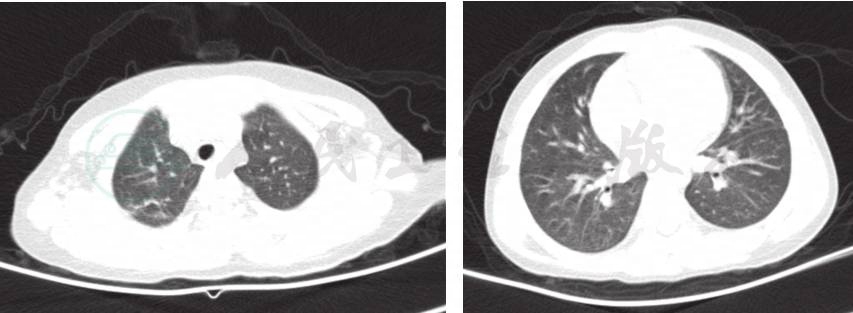
图2 入院后肺CT提示:右肺上叶尖段及左肺下叶基底段少许炎症
以上辅助检查结果高度疑似“高IgM综合征”,予患儿及其父母行相关基因检测。患儿入院第2天仍有高热,予患儿抗生素升级为头孢吡肟,且给予静脉注射丙种球蛋白,在行骨髓穿刺术检查排除血液系统疾病后给予肌注重组人粒细胞刺激因子以提升中性粒细胞,同时口服铁剂,同服复方新诺明防治非典型菌感染;3天后患儿热退,咳嗽减轻,5天后复查血常规:WBC 3.2×109/L,NE%23.4,N 0.75×109/L,RBC 4.42×1012/L,HGB 97g/L,HCT 31.7%,MCV 71.7fl,MCH 21.9pg,MCHC 306g/L,PLT 513×109/L。CRP 32.2mg/L。PCT 0.099ng/L。粒细胞数有所提升,感染指标均较前明显好转。将抗生素降级为头孢呋辛,继续抗感染治疗5天后复查血常规:WBC 6.4×109/L,NE%41.2,N 2.64×109/L,RBC 4.26×1012/L,HGB 98g/L,HCT 31.8%,MCV 71.9fl,MCH 22.0pg,MCHC 307g/L,PLT 463×109/L。CRP 7.9mg/L。PCT 0.062ng/L。免疫球蛋白:IgG 6.0g/L,IgA<0.25g/L,IgM 1.3g/L;患儿无发热,无咳嗽,顺利出院。嘱患儿于笔者医院儿科门诊定期检测血常规及免疫球蛋白,适时输注丙种球蛋白。患儿出院约1.5个月后基因检测回报,患儿确诊为X连锁高IgM综合征,存在CD40L基因突变,其母亲为该致病基因携带者,建议予患儿行骨髓移植彻底治愈本病,家长未同意;患儿在定期输注丙种球蛋白后未再发生严重感染。
1.X连锁高IgM综合征。
2.急性支气管肺炎。
3.脓毒症。
4.粒细胞缺乏症。
5.小细胞低色素性贫血。
高IgM综合征(hyper-immunoglobulin M syndromes,HIGM)是一种较罕见的原发性免疫缺陷病,20世纪60年代由Asselain和Rosen等首次报道。其主要特点为反复感染,血清IgG、IgA和IgE明显降低,IgM水平正常或升高,B淋巴细胞数正常。
HIGM患者主要表现为反复细菌感染,如上呼吸道、肺部细菌感染和中耳炎等,频繁发生某些机会性感染,如卡氏肺囊虫、小隐孢子虫、弓形虫等感染。自身免疫性疾病及恶性肿瘤发病率明显升高,各种胃肠肿瘤、肝细胞癌、腺癌、胆管癌均可发生。HIGM患者体检可见扁桃体、颈部淋巴结大,肝脾大也可出现。血清学检查免疫球蛋白总量可正常,IgM增高或正常,IgG、IgA、IgE大多明显降低或缺如。由于抗体生成存在缺陷,易罹患胞外细菌感染,主要表现为反复发作的球菌性肺炎、中耳炎、鼻窦炎、肺炎,最终导致支气管扩张,免疫球蛋白补充疗法对预防出现上述症状有较好的效果。卡氏肺孢子虫肺炎(PCP)是最常见的机会性感染,其发生率高达40%。慢性隐孢子虫病也是常见的机会性感染之一,可出现症状性的慢性肠道隐孢子虫病,主要表现为顽固性腹泻,可导致体质量下降,甚至死亡。胆道系统感染也是HIGM常见的机会性感染之一,主要表现为肝功能异常(ALT升高为主),随病情进展,可演变为硬化性胆管炎,甚至转变为胆管癌。慢性肝损伤占HIGM的50%,是导致很多病例死亡的主要原因。巨细胞病毒感染也是HIGM常见的机会性感染,也是硬化性胆管炎中常见的病原体。中性粒细胞减少在CD40 L缺陷的男童中最常见,其发生率高达50%。中性粒细胞减少可能是暂时的,也可能持续存在,甚至终生存在,其发生机制尚未阐明,部分病例可检测到抗中性粒细胞抗体,髓系细胞前体表达CD40和CD40 L对于刺激髓系发育有重要意义。研究表明,大剂量IVIG对于纠正中性粒细胞减少有很大作用,但欧洲大样本研究表明这种方法仅对半数病例有效,可用粒细胞集落刺激因子(G—CSF)纠正中性粒细胞减少。淋巴系统增殖反应在AID缺陷患儿最常见,占1/2~2/3,最常见的为淋巴结和扁桃体大,脾大不多见。淋巴结和扁桃体活检显示生发中心增生,免疫球蛋白替代治疗可降低其发生率。恶性疾病在CD40 L缺陷较为多见,胆道系统和肠道恶性肿瘤最常出现,其次为神经内分泌肿瘤,淋巴瘤并不多见。自身免疫性疾病在CD40 L/CD40缺陷较多见。20%的AID缺陷发生自身免疫性疾病,主要包括自身免疫性血细胞减少、关节炎和肝炎。
HIGM的实验室检查主要表现为血清IgG、IgA和IgE明显降低,IgM水平正常或升高。临床表现为反复感染、血清IgG、IgA和IgE明显降低,IgM水平正常或升高的PID较多,需与CVID、XLA等疾病相鉴别,另外还需要考虑:①实验误差:若仅发现IgM升高,而临床症状无HIGM的表现,则有可能为实验室操作误差,需重新检测。②某些肠道疾病或肾脏疾病:由于低相对分子质量蛋白的丢失(IgG和IgA),可出现IgG和IgA降低,而IgM正常的类似HIGM表现。另外,需要指出的是并非所有IgM升高均为HIGM,并且不同年龄段IgM水平存在差异。
治疗方面:①抗感染对症支持治疗:急性感染期,可应用抗细菌、病毒及真菌药物,对于CD40 L缺陷所导致的粒细胞缺乏,可用G-CSF(非格司亭)升高粒细胞。②IVIG补充疗法:HIGM一旦确诊,即应静脉注射 IVIG,IVIG的推荐剂量为400~600mg/kg,每3、4周1次。可纠正体液免疫缺陷,但对于机会性感染的预防作用尚存在争议。③对CD40 L缺陷,有学者尝试注入可溶性CD40 L,但由于CD40不仅表达于免疫细胞,还表达于其他细胞系,此方法特异性不强,在纠正B淋巴细胞产生抗体功能的同时,可能导致其他细胞系功能紊乱,目前不推荐使用。④造血干细胞移植:目前,造血干细胞移植仍是治愈 HIGM的最好方法。2006年,国内首例XHIGM患儿骨髓移植成功。目前该患儿已停用抗排斥药物,无感染,血清IVIG水平和造血功能已恢复健康人水平。⑤基因疗法:近年来,有学者尝试应用基因疗法治疗HIGM。动物实验表明CD40 L敲除小鼠在CD40 L基因重诱导后,CD40 L重新表达,但却引起淋巴细胞增殖性疾病。因此,推测要实现该基因的精确表达,不仅包括该基因结构的表达,还应包括调控该基因蛋白的表达。因此,HIGM基因疗法尚在试验阶段。
(王 佳 韩晓华)









