 去看看
去看看

一、概述
自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA)是由于机体免疫功能紊乱,产生红细胞自身抗体,通过抗体或补体途径介导红细胞破坏增多,超过骨髓红系代偿增生能力所致的一组溶血性贫血的统称。AIHA于整个儿童期均可发病,发病率无明显种族差异。国外资料显示儿童AIHA年发病率估计为(0.81~2)/10万。为规范我国儿童AIHA诊治,经国家卫生健康委员会儿童血液病专家委员会专家讨论,制定本诊疗规范。
二、AIHA病因与分型
(一)病因与分型
1. 依据病因分为原发性和继发性AIHA两类。
(1) 原发性AIHA:也称为特发性AIHA,这类AIHA患儿无明确可引起溶血的基础疾病。原发性AIHA占儿童AIHA病例的40%~50%,大多由温抗体型自身抗体导致。
(2)继发性AIHA:这类AIHA患儿有可引起溶血的基础疾病。儿童AIHA病例系列研究显示:超过50%具有免疫性疾病,约10%继发于感染。所有AIHA患儿都需要考虑存在潜在全身性疾病的可能;尤其青少年和2岁以下儿童,以继发性AIHA更为多见,病程更为缓慢。较常见继发原因如下:
①自身免疫性疾病:全身性自身免疫性疾病是AIHA常见的继发性病因,尤其是大龄儿童,与AIHA相关的自身免疫性或炎症性疾病包括系统性红斑狼疮(SLE)、干燥综合征、硬皮病、幼年特发性关节炎、皮肌炎、白癜风、溃疡性结肠炎、自身免疫性肝炎、糖尿病和自身免疫性甲状腺炎等。异基因造血干细胞移植后自身免疫排斥也可导致AIHA发生。
②免疫缺陷:原发性免疫缺陷病(Primary immunodeficiency,PID)可因免疫功能紊乱而引起继发性AIHA,AIHA甚至可以是PID确诊前的初始表现,特别是普通变异型免疫缺陷病(common variable immunodeficiency,CVID)和湿疹血小板减少伴免疫缺陷综合征(Wiskott-Aldrich syndrome,WAS)。自身免疫性淋巴细胞增生综合征(autoimmune lymphoproliferative syndrome,ALPS)由于遗传性淋巴细胞凋亡缺陷造成免疫失调被归为PID,可因自身反应性淋巴细胞的扩增和增殖引起AIHA。获得性免疫缺陷症,如HIV感染,可由于多克隆B淋巴细胞激活和T淋巴细胞免疫调节功能障碍,引起继发性AIHA。
③恶性肿瘤:AIHA也可继发于多种恶性肿瘤,甚至发生在恶性肿瘤诊断前。常见肿瘤类型包括霍奇金淋巴瘤、急性白血病、骨髓增生异常综合征等。
④感染:是AIHA的另一重要原因,常见感染源包括肺炎支原体、EB病毒、麻疹病毒、水痘病毒、腺病毒、腮腺炎病毒和风疹病毒。此类AIHA的自身抗体多为与红细胞I/i多糖抗原特异性结合的IgM型自身抗体,临床多表现为冷凝集素病(cold agglutinin disease,CAD)。而多数阵发性冷性血红蛋白尿(paroxysmal cold hemoglobinuria,PCH)患儿,发病前多有病毒感染表现,但具体病原体一般不能明确。
⑤药物:药物可引起红细胞自身抗体的产生,但需要药物形成半抗原或需要药物与红细胞形成抗原抗体复合物。引起儿童AIHA的常见药物有青霉素类、头孢菌素类、四环素、红霉素、丙磺舒、对乙酰氨基酚和布洛芬等。
⑥Evans综合征:当患儿同时存在AIHA和免疫性血小板减少症(immune thrombocytopenia,ITP)时,则被称为Evans综合征,某些Evans综合征可伴有自身免疫性中性粒细胞减少。部分患者最初表现为孤立性AIHA,在数月甚至数年后再出现其他免疫性血细胞减少;另一些患儿最初发生ITP,随后发生AIHA。Evans综合征占儿科AIHA病例的15%~30%。与孤立性AIHA相比,Evans综合征患儿的AIHA多为继发性,治疗更难,临床病程往往呈现慢性复发性。
2. 依据自身抗体与红细胞结合所需的最适温度将AIHA分为温抗体型、冷抗体型AIHA(包括CAD及PCH)。
(1)温抗体型AIHA:是儿童原发性AIHA的最常见类型,占60%~90%,抗体类型常为IgG型。这些抗体在37℃条件下优先结合红细胞,导致血管外溶血(主要发生于脾脏),继而引起贫血、黄疸,偶尔引起脾肿大。这类与红细胞的最适反应温度为35~40℃的自身抗体称为温抗体。某些情况下,IgG型自身抗体数量和密度足以固定补体,从而同时引起血管内溶血。
(2)冷凝集素病(CAD):儿童CAD相对少见,约占10%,最常发生在肺炎支原体或EB病毒感染后。CAD相关IgM自身抗体在低温条件下(0~4℃)与红细胞I/i抗原结合并固定补体,进而介导血管内溶血。这类与红细胞的最适反应温度在30℃以下特别是4℃的自身抗体称冷抗体。此类抗体亦可通过肝脏巨噬细胞介导红细胞免疫性破坏和清除,引起血管外溶血性贫血。
(3)阵发性冷性血红蛋白尿症(PCH):是一种几乎仅发生于儿童的AIHA,最常发生在病毒感染后。PCH的特征为IgG自身抗体在低温下更容易结合、有效固定补体,复温时引起血管内溶血。
3. 抗体类型与原发性和继发性AIHA的关系。
(1) 温抗体型AIHA:分为原发性(特发性)和继发性AIHA。继发性因素包括:自身免疫或炎症性疾病(例如SLE、Sjogren综合征、硬皮病、幼年型特发性关节炎、皮肌炎、白癜风、溃疡性结肠炎、自身免疫性肝炎、糖尿病、自免性甲状腺炎);Evans综合征;PID(例如CVID、WAS);获得性免疫缺陷病(例如HIV感染);恶性病(例如急性白血病、淋巴瘤);感染和移植后状态。
(2) CAD:包括原发性(特发性)和继发性CAD。继发性因素包括:感染(例如支原体肺炎、EB病毒感染);恶性病(例如淋巴瘤);自身免疫性疾病;造血干细胞移植后。
(3) PCH:包括原发性(特发性)和继发性PCH,继发性多发生于病毒感染后。
三、临床表现
AIHA的症状和体征与其他类型溶血性贫血相同。症状严重程度取决于溶血严重程度和发生速率。轻症者可无症状(仅化验显示轻度贫血),严重者可存在重度~极重度贫血,甚至危及生命。
(一)症状
大多数AIHA患儿有贫血相关的症状和体征,如无力、乏力、呼吸急促、头晕、苍白和/或溶血相关症状和体征,如黄疸、茶色尿、酱油色尿等。其他非特异性症状包括腹痛或发热。CAD患儿最常见的表现可有寒冷后红细胞凝集相关症状如肢端发绀(即指尖、脚趾等肢端最远端和耳鼻皮肤变为深紫色到灰白色);温暖时症状消失。排出葡萄酒色、可乐色和黑色血红蛋白尿常提示有冷抗体型AIHA导致的血管内溶血,如CAD或PCH。如果贫血缓慢发生,则心血管系统可以良好代偿;而急速发生极重度贫血时则会导致氧供不足的心血管失代偿表现。
(二)病史
须询问是否有类似发作史、近期急性感染史、服药史和是否有免疫性疾病病史和/或家族史。
(三)查体
可能发现苍白和黄疸,结膜和手掌尤为明显。患者常有心动过速和收缩期血流杂音;如急性严重AIHA发作,可出现心力衰竭。查体可触及肿大的肝脏和脾脏。
四、实验室检查
(一)初始实验室检查
全血细胞计数(complete blood count,CBC)、红细胞指数、网织红细胞计数、外周血涂片、直接抗人球蛋白试验(Direct antiglobulin test,DAT,直接Coombs试验)、尿液分析(试纸尿干化学检测法和显微镜下观察)、血尿素氮(blood urea nitrogen,BUN)和肌酐、间接胆红素、乳酸脱氢酶(lactate dehydrogenase,LDH)、天冬氨酸转氨酶(AST)。如果有血红蛋白尿、肺炎支原体感染、寒冷暴露下肢端发绀等,还应测定冷凝集素滴度。疑诊新生儿溶血病或血型不合溶血性贫血时可加查间接抗球蛋白试验。
1.全血细胞计数:贫血常为重度,伴有网织红细胞百分比和绝对数升高。白细胞计数和血小板计数通常正常或升高。
2.血涂片检查:温抗体型AIHA患儿外周血涂片上常可见球形红细胞;而存在冷凝集素时,红细胞可能聚集成小团块通过自动计数仪器,导致MCV假性增大;当网织红细胞从骨髓中释放以代偿加速的红细胞破坏时,常可观察到红细胞的嗜多染性以及Howell-Jolly小体和有核红细胞。
3.直接抗球蛋白试验(DAT):是AIHA的确诊试验,能识别红细胞表面的抗体和/或补体类型,以半定量形式评分。DAT结果的阳性强弱一般与溶血严重程度相关。常规DAT试验检测IgG型、IgM型和/或C3d型抗体。少于5%的患儿尽管有临床温抗体型AIHA证据,由于红细胞表面IgG的量低于标准DAT能检测到的阈值而使结果呈阴性。罕见情况下,患儿也可能因仅存在IgA型自身抗体,甚至仅存在温反应性IgM自身抗体,从而导致常规DAT呈现假阴性。此时需采用特定的研究试剂和技术,如微柱凝胶卡或流式细胞仪以助鉴别。对于冷凝集素介导性AIHA,DAT结果通常为抗C3d阳性,抗IgG阴性。发生这种情况时,应检查患儿血清是否有冷反应性Donath-Landsteiner抗体(D-L抗体)存在。D-L抗体是IgG型冷热溶血素,在0~4℃时与红细胞结合,并吸附补体,但并不溶血;在复温至30~37℃时诱发溶血(表1)。
4.间接抗球蛋白试验(IAT):DAT的目的是检查红细胞表面的不完全抗体,而IAT试验的目的是检查血清中是否存在游离抗体。通常用于检测疑诊新生儿溶血病患者母体抗体以及因红细胞不相容的输血而产生的血型抗体。
5.冷凝集素滴度:血清冷凝集素滴度是指寒冷暴露下(0~4℃)仍可看见红细胞凝集的血清样本最大稀释度。几乎所有正常人都有低滴度的冷凝集素(<1:40),但感染后的冷凝集素病时,该滴度通常高于1:256;而淋巴瘤相关性冷凝集素病时,滴度更高,通常高于1:2000。
6.尿液分析与肾功能检查:血管内溶血的患儿尿液表现为血红蛋白尿(潜血阳性但镜检无红细胞)。慢性血红蛋白尿会导致尿液含铁血黄素聚集,表现为尿沉渣检测时细胞铁染色阳性(尿Rous试验阳性)。而血管外溶血(如温抗体型AIHA)患者的尿液分析结果仅有尿胆原升高。溶血可引起肾功能不全,因此初始评估时应检测尿素和肌酐。
7.溶血的血清标志物:溶血的血清标志物异常可帮助确定是否有溶血性贫血,但这些检测对AIHA不具特异性:总胆红素增高,以非结合形式(即间接胆红素)为主;血清LDH和AST浓度升高,尤其是血管内溶血时,而ALT和其他肝酶基本正常;多数AIHA病例中,血清结合珠蛋白(可结合血浆游离血红蛋白)水平较低或不存在(小婴儿合成结合珠蛋白能力弱、且结合珠蛋白也是急性期反应物,存在炎症或感染时其浓度可能升高。因此,患者小于18个月龄时,不建议常规检测结合珠蛋白水平)。
五、诊断
(一)诊断依据
同时存在溶血性贫血证据(红细胞破坏证据和代偿增生证据)和红细胞抗体证据(DAT阳性结果)。
(二)温抗体型和冷抗体型AIHA的诊断标准
1. 温抗体型AIHA
当以下情况均存在时:①有溶血性贫血(贫血、间接胆红素水平高、LDH水平高、结合珠蛋白水平低)证据;②DAT结果为抗IgG阳性,红细胞上可能存在补体;③如果外周血涂片显示球形红细胞且有网织红细胞增多和红细胞平均血红蛋白浓度升高,则支持诊断为该病,但不是诊断必须。
2. CAD
当以下情况均存在时:①溶血性贫血(贫血、间接胆红素水平高、LDH水平高、结合珠蛋白水平低);②DAT结果为补体C3阳性,IgG阴性;③外周血涂片上红细胞聚集和较高的冷凝集素滴度支持诊断为该病,但不是诊断必须。
3.PCH
当以下情况均存在时:①溶血性贫血(贫血、间接胆红素水平高、LDH水平高、结合珠蛋白水平低);②DAT结果为补体C3阳性;③冷热溶血试验阳性。
(三)确定原发、继发性
1. 评估有无继发性原因。所有新诊断的AIHA患儿都应筛查继发性病因,并常在治疗前开展,如抗核抗体、免疫球蛋白定量检测、血清学检查以判断有无肺炎支原体和EB病毒感染(仅针对冷凝集素病患儿)、回顾患儿的用药情况。但对于需要尽快治疗、危及生命的重度贫血儿童,则以治疗优先。
由于许多情况下继发性疾病在诊断出AIHA后才显现出来,因此对于病情迁延,即使暂未发现继发性病因而诊断为原发性AIHA患儿,也应定期重复筛查。
2. 如果患儿存在如复发性感染病史、免疫缺陷或自身免疫性疾病家族史、其他血细胞减少(中性粒细胞减少和/或血小板减少)、淋巴结肿大和/或器官肿大病史、体格检查或实验室检查发现异常情况,提示潜在恶性肿瘤、免疫缺陷或自身免疫性疾病时,就需进行相关检查以明确病因。可根据临床表现选择淋巴细胞亚群检测、HIV检测、骨髓穿刺和活检、胸片、腹部超声检查或CT等影像学检查、使用流式细胞仪筛查有无ALPS、通过抗双链DNA抗体、C3和C4筛查有无SLE等。
3. 存在同种异体抗体检测:对于在子宫内或通过之前的输血暴露于外源红细胞的AIHA患者,应筛查IAT判断患者血浆中是否存在同种异体抗体。若检出同种异体抗体,则在寻找相匹配的血液时,需要进行扩大的血液分型或基因分型。若存在这些抗体但又未发现,可导致患者发生严重的输血反应(表2)。
六、鉴别诊断
通常所说的AIHA是指原发性AIHA,这些患儿通常具有溶血性贫血的表现、体征及实验室证据(例如红细胞破坏增多及骨髓红系代偿增生的证据),因此,非免疫性原因导致的溶血性贫血需要重点鉴别。其次,儿童的AIHA以继发性AIHA为主,诊断过程中尚需要注意明确是否有继发因素(参看病因与分型)。主要的鉴别诊断如下:
(一)遗传性球形红细胞增多症(HS)及其他先天性溶血性贫血
HS很易与AIHA混淆,因为两种疾病均存在外周血球形红细胞和网织红细胞增多,骨髓中粒红比倒置,且可能均有脾大、红细胞渗透脆性升高。但前者可能有家族史,伴胆囊结石,而后者通常没有。后者DAT通常为阳性,而前者为阴性。此外,遗传性椭圆形红细胞增多症、红细胞葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症、地中海贫血等其他先天性溶血性贫血,由于同样具有溶血的表现及体征而易与AIHA相混淆,DAT同样是区分AIHA与其他原因导致的溶血性贫血的主要依据。
(二)其他类型贫血
血栓性微血管病(例如溶血尿毒综合征、血栓性血小板减少症、弥散性血管内凝血等)、机械瓣膜病等均可通过机械力量破坏红细胞导致溶血性贫血的发生。此类贫血外周血涂片中可见到破碎红细胞,DAT检测为阴性。巨幼红细胞性贫血、甲基丙二酸血症等因维生素B12代谢异常,可导致巨大红细胞出现,骨髓原位溶血,此时可伴有总胆红素升高,以间接胆红素升高为主,网织红细胞比例或绝对值升高。但前者血清维生素B12、叶酸水平下降,后者血尿筛查可见甲基丙二酸血症或尿症,两者均DAT试验阴性,与AIHA不同。
纯红细胞再生障碍性贫血等先天性骨髓衰竭综合征、获得性再生障碍性贫血或暂时性成红细胞减少症等也可以引起贫血,但这些贫血是由于红细胞生成减少引起的,相比较于溶血性贫血,这些疾病的网织红细胞计数通常较低。
(三)黄疸
儿童出现黄疸时需要具体分析何种胆红素水平升高。在溶血性贫血中,通常是间接胆红素升高,而肝脏疾病时通常为直接胆红素升高。
1. 肝病:氨基转移酶和凝血因子检测有助于评估肝脏功能。在溶血性贫血中,间接(非结合)胆红素水平升高,而肝脏疾病直接(结合)胆红素水平升高。
2. Gilbert综合征:又称为体质性肝功能不良性黄疸,属一种较常见的遗传性非结合胆红素血症,临床表现特点为长期间歇性轻度黄疸,无贫血症状。Gilbert综合征为常染色体显性遗传性疾病,患者主要为青少年,男性多见。
(四)深色尿
尿液中出现血红蛋白的深色尿可与血尿或肌红蛋白尿相混淆。此时需要做尿液及生化分析以鉴别。血尿的尿液镜检中可见到大量红细胞,而血红蛋白尿中通常没有红细胞。肌红蛋白尿的尿液分析与血红蛋白相似,但肌红蛋白通常发生于快速大量的肌肉分解,血生化中肌酶水平明显升高,血红蛋白尿的患者通常不会有肌酶水平的升高。血红蛋白尿提示存在病理状态的血管内溶血,包括AIHA、红细胞G6PD缺乏症、阵发性睡眠性血红蛋白尿等。可以通过检测G6PD酶活性、CD55/CD59水平加以鉴别。
(五)AIHA鉴别诊断流程图
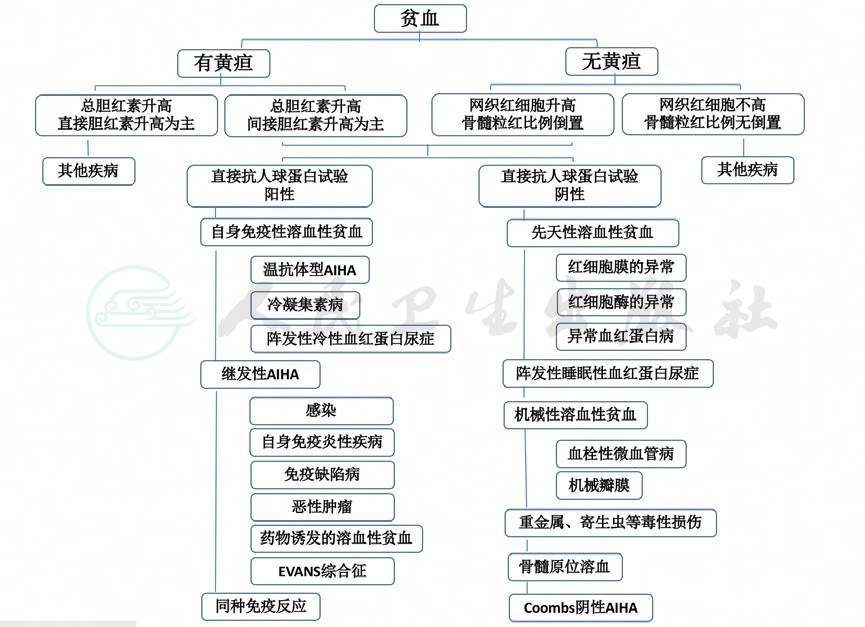
七、治疗
(一)一般处理
AIHA患儿的最佳治疗策略需要结合贫血程度、发病的急慢性、症状和体征以及自身抗体的特点。如果贫血不严重且没有心衰表现,则AIHA患儿可在门诊治疗;而急性起病时可能需要住院,进行诊断性评估、严密监测患者并对危重症患儿开展抢救。
继发性的AIHA需要迅速脱离接触因素(如药物)、控制原发病(如感染、肿瘤等),治疗才有好的效果。
(二)支持治疗
AIHA由于存在自身抗体,增加了交叉配血难度,增大了同种抗体致溶血性输血反应的危险,因此应尽量避免或减少输血。
但如果贫血已经危及生命时也应输血。输血时机应根据贫血程度、有无明显症状、发生快慢而定。抢救时不强调应用洗涤红细胞,交叉配血不完全相合时,可选用多份标本交叉配血中反应最弱的输注,并需要缓慢输注,密切观察有无输血反应。此时输注的供体红细胞在AIHA患者体内存活时间与患者自身红细胞相近,但即使输注的红细胞在循环中仅能存在较短的时间,仍可帮助改善贫血症状和体征。输血前加用糖皮质激素可以减少或减轻输血反应的发生。
对于急性溶血性贫血患儿,出现严重症状时能排除同种抗体者,需要立即输注红细胞;对于慢性贫血者,HGB在70g/L以上可以不输血;HGB在50g/L~70g/L时,如果有不能耐受的症状时可以适当输血;HGB在50g/L以下时应考虑输血。存在冷抗体型自身抗体的患儿,更易发生重度溶血性输血反应。对于此类患者,降低溶血性输血反应可能性的策略包括:输注前和过程中加热血制品至37℃;以缓慢的速度开始输血,并定期检测血浆和尿液样本有无游离血红蛋白。在罕见的情况下,输血使溶血加重时,可伴显著的血红蛋白血症及血红蛋白尿,应该充分补液和碱化以防止肾功能衰竭。
PCH患儿通常有突发但自限性溶血,治疗的主要措施是使患者保暖及避免接触冰冷液体。大多数冷凝集素(IgM)的CAD患儿是由肺炎支原体或EB病毒感染所致。贫血往往很轻,大部分病例不需要药物治疗。与PCH一样,其管理包括使患者保暖及避免接触冰冷液体。治疗基础病因(如对支原体感染患者应用抗生素)也是另一重要方面。
(三)药物治疗
1. 温抗体型AIHA:通常易反复和呈现慢性过程,不经治疗常不能缓解,需给予药物治疗。
(1) 一线治疗
糖皮质激素是温抗体型AIHA的一线治疗。推荐在无糖皮质激素禁忌情况下应用。对于温抗体型AIHA,糖皮质激素常在24h~48h内起效,治疗反应率为50%~80%,但减量过快或突然停用时易复发。PCH患儿通常有自限性溶血,但可能需要短期使用糖皮质激素以减少溶血和改善贫血。少数情况下,糖皮质激素对CAD可能有效。
①初始剂量取决于贫血的程度:重度贫血患儿需住院治疗,在最初的24~72h,每6~8h静脉给予甲泼尼龙(1~2)mg/kg。严重者也可以采用冲击剂量,如甲泼尼龙30mg/(kg·d),最大剂量为1g/d,连续3d;随后改为常规剂量维持。轻中度贫血患儿可以接受口服强的松治疗,剂量(1~2)mg/(kg·d),最大剂量为60mg/d。
②减量可先快后慢:糖皮质激素用至红细胞比容大于30%或者HGB水平稳定于100g/L以上才考虑减量,并根据患者耐受情况在2~6个月内逐渐减量,例如每月减少2.5~10.0mg。后期应将糖皮质激素缓慢减量,直到血红蛋白、网织红细胞计数、LDH和结合珠蛋白正常。
③治疗过程中需严密监测并发症:儿童长期应用糖皮质激素可引起多种并发症,包括体质量增加、生长受损、精神和认知症状、高血压、高血糖、骨质疏松、股骨头坏死和白内障等。
④其他治疗和监测:需要注意碱化利尿(严重贫血时不强调水化。血管内溶血时需要适度碱化。血管外溶血时无需碱化)、利胆去黄,并注意电解质平衡。定期临床评估及监测血红蛋白水平、网织红细胞计数、LDH和DAT,监测频率取决于疾病严重程度和治疗。实验室检测通常首先每周监测1次;随着临床情况的稳定,检查的间隔时间可以逐渐延长。通常监测至少1年。如在完全缓解后,DAT仍为阳性,则需进一步寻找继发性病因,如Evans综合征或系统性红斑狼疮。
⑤治疗反应:
预期反应:约80%的患者应用糖皮质激素治疗会获得初始反应。研究显示,大多数患儿在1个月内出现血红蛋白水平恢复,但72%的患儿接受了至少6个月的治疗。
复发:AIHA患者常复发,15%~40%的患者在获得初始反应后最初6个月到1年期间发生复发。复发治疗通常是重新开始既往能够有效维持患者缓解的最低剂量泼尼松治疗。
糖皮质激素依赖:是指对于减量频繁复发或服用低剂量泼尼松(每日≤0.2mg/kg)不能维持缓解的患者。
⑥疗效标准:
痊愈:继发于感染者,在原发病治愈后,AIHA也治愈。表现为无临床症状、无贫血、DAT阴性。CAD者冷凝集素效价正常,PCH者冷热溶血试验阴性。
完全缓解:临床症状消失,红细胞计数、HGB水平和网织红细胞百分比均正常,血清胆红素水平正常。DAT阴性。部分缓解:临床症状基本消失,HGB>80g/L,网织红细胞百分比<4%,血清胆红素<34.2μmol/L。DAT阴性或仍然阳性但效价较前明显下降。
无效:仍然有不同程度贫血和溶血状态,实验室未达到部分缓解标准。
此外,一线治疗还包括和糖皮质激素联合使用的静脉免疫球蛋白(intravenous immunoglobulin,IVIG)冲击治疗,对温抗体型AIHA有效,剂量为0.4g/(kg·d),连续5d静脉输注;或1g/(kg·d),连续2d冲击治疗。对于复发或难治性AIHA,可以考虑多次使用。IVIG对成人AIHA是一种非常有吸引力的选择,但大多数患儿单独应用IVIG无反应,即使产生反应,疗效也通常短暂。
(2)二线治疗
以下情况需要予二线治疗:1~2个月内糖皮质激素治疗无反应的患者,不耐受糖皮质激素逐渐减量、每日维持剂量>1mg/kg的泼尼松剂量,糖皮质激素依赖,其他禁忌或不耐受糖皮质激素治疗,AIHA复发。
①利妥昔单抗:静脉给药,每剂375mg/m2,每周1次,应用2~4周,反应率为60%~85%。也有报道显示小剂量利妥昔单抗(100mg/次,每周1次,连续4次),可降低经济负担及不良反应,而不降低疗效,但临床数据及随诊时间有限。利妥昔单抗应用时需要注意过敏反应,大多数发生于首次输注时,治疗前可采用对乙酰氨基酚和抗组胺药预防。严重不良反应包括重度的输液相关反应、乙肝病毒感染再激活、重度的皮肤黏膜反应和进行性多灶性白质脑病。应在开始利妥昔单抗治疗前对所有患者根据当地指南进行乙肝病毒(HBV)的筛查,不应对处于乙肝活动期的患儿使用利妥昔单抗治疗。对于HBV血清学检测阳性的患者,在开始接受治疗前应咨询肝病专科医生的意见,同时应对其开展监测以预防HBV再激活的发生。
②脾切除:脾切除是慢性或顽固性AIHA患儿有效治疗手段;约2/3的患儿有短期改善,通常在手术后2周内改善明显。幼儿脾切除后存在荚膜细菌所致脓毒症风险,因此3岁以下患儿应避免进行脾切除,最好延迟至6岁以后进行。准备脾切除的患者,应该在术前适当的时间进行抗肺炎链球菌、脑膜炎奈瑟菌和B型流感嗜血杆菌的免疫。已进行了脾切除的患儿酌情予长效青霉素预防性治疗,并应嘱患者在出现发热时立即就医。
(3) 难治性AIHA:对于糖皮质激素、利妥昔单抗和/或脾切除治疗失败的难治性AIHA、以及在应用利妥昔单抗和/或脾切除后仍依赖类固醇治疗的难治性AIHA患儿,可能需要更积极的治疗。此时虽然尚有一些治疗选择,但都不如糖皮质激素、利妥昔单抗和脾切除有效。
①硫唑嘌呤和6-巯嘌呤:6-巯嘌呤及其前体药物硫唑嘌呤是免疫抑制药物,主要影响辅助性T淋巴细胞功能,从而减少自身抗体的合成并发挥“类固醇助减”作用。这两种药物可能都需要2~3个月以上的治疗才能出现临床反应。应用过程中,需注意出现骨髓抑制、肝功损害、皮疹等副作用可能。
②环孢素A:主要影响T淋巴细胞功能。建议起始口服剂量为每日5mg/kg,每12h 1次,维持血药浓度(谷浓度)100~200μg/L。由于环孢素A需要达到有效血药浓度后才起效,建议初期与糖皮质激素联用。环孢素A不良反应有齿龈增生、多毛、高血压、胆红素增高、肾功能受损等。
③吗替麦考酚酯、他克莫司和西罗莫司:这些免疫抑制剂可用于复发、难治AIHA患者,它们的应用可能发挥“类固醇助减”作用,从而减少维持缓解所需糖皮质激素的剂量。目前关于这些药物用于AIHA治疗的数据较少。
④细胞毒制剂:可以减少自身抗体生成的细胞毒制剂包括长春新碱、长春花碱和环磷酰胺。但这些药物一般具有骨髓抑制性及致突变作用,在儿童患者中应谨慎使用。
⑤血浆置换:血浆置换对冷凝集素IgM介导的AIHA效果较好(37℃时80%IgM型抗体呈游离状态);但对温抗体型AIHA效果不佳,且置换常带入大量补体。
⑥造血干细胞移植:AIHA患者进行成功的造血干细胞移植已有报道、仅用于其他所有治疗均失败的重度AIHA患者。
(四)继发性AIHA
对于某些原因的继发性AIHA,治疗时还需考虑:
1.Evans综合征:许多Evans综合征患者采用糖皮质激素治疗无效,需开展二线治疗。吗替麦考酚酯和西罗莫司可能有效。ALPS最有可能是Evans综合征的患儿免疫系统失调的基础病因,因此,表现为Evans综合征的所有患儿都必须筛查有无ALPS。
2. 免疫缺陷:具有基础免疫缺陷患者的AIHA治疗与原发性AIHA相似,大多数患者对糖皮质激素治疗有反应。然而,具有基础免疫缺陷的患者,特别是CVID,在脾切除或利妥昔单抗治疗后可发生重度感染。因此,对这类患者需密切监测感染性并发症发生可能。
3. ALPS:ALPS患者发生AIHA时,通常表示自然病程的转变,并且需要开始或加强治疗。有自身免疫表现的患者比仅有淋巴组织增生的患者通常更难对免疫抑制治疗产生反应。这些患者可能需要免疫抑制剂联合免疫调节剂治疗,如糖皮质激素、利妥昔单抗、IVIG、吗替麦考酚酯和西罗莫司。
4. 药物诱发的AIHA:对于疑似药物诱发的AIHA患者,必须停用可疑药物。很多情况下,需应用糖皮质激素以减慢溶血速度;如果患者临床情况恶化,也可考虑血浆置换。重度快速进展性贫血患者可能需要静脉给予大剂量甲泼尼龙(每日30 mg/kg,治疗3d;日均最大剂量1g),随后口服泼尼松且根据疗效逐渐减少每日剂量。
(五)儿童AIHA的治疗流程图
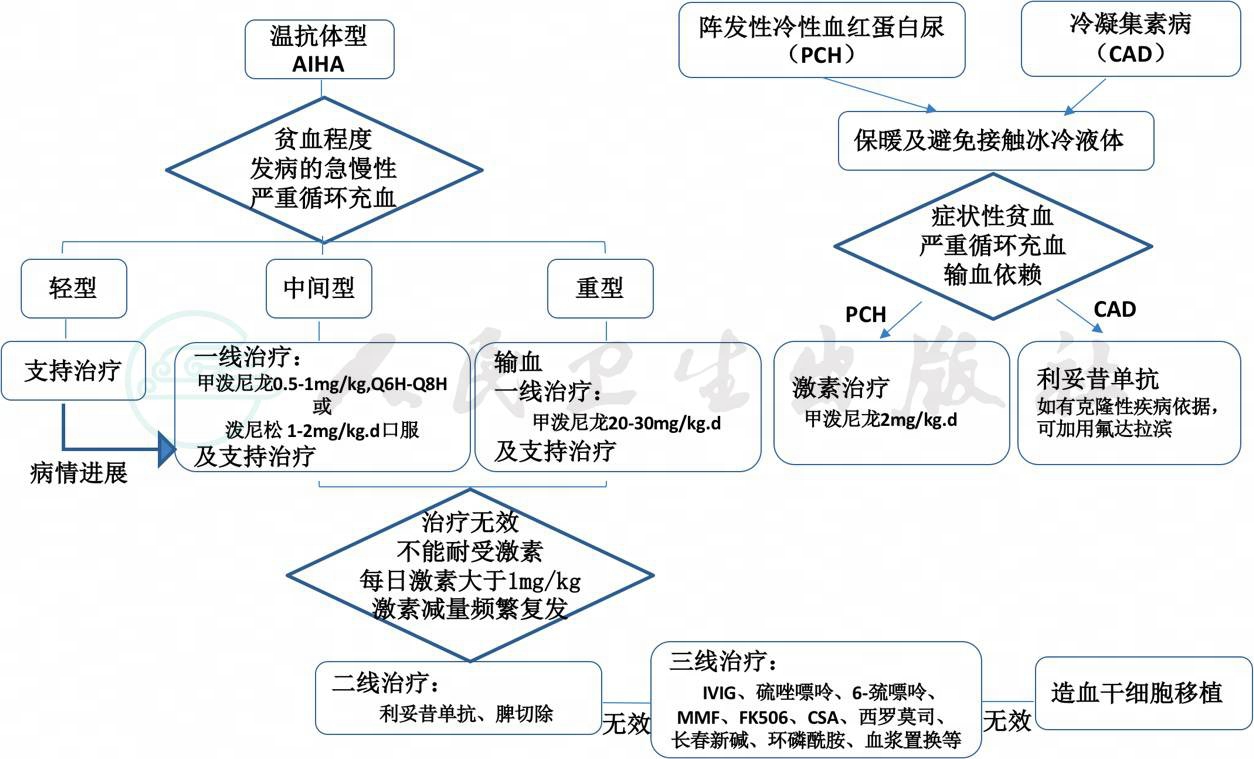
八、预后
据报道,在1994~2014 年间进行治疗的AIHA患儿病死率为3%~4%,死亡的主要病因是脾切除患者发生过于严重的脓毒症、Evans综合征患者发生灾难性的出血(同时并发ITP)以及基础疾病(恶性肿瘤)。
PCH和继发于肺炎支原体或EB病毒感染的CAD患儿可急性起病、病情较重,但大多数患者在起病后数周至数月内可获得完全缓解,病程常具有自限性,预后好。相比之下,IgG介导的温抗体型AIHA患儿常为慢性病程,以间断复发为特点。
九、转诊条件
(一)从上级医院转诊到下级或基层医院
1. 诊断及疾病状态明确,暂不需要进一步特殊检查;
2. 临床状态稳定,无心力衰竭、肾功能不全等并发症,血红蛋白水平稳定或逐步回升;
3. 治疗方案确定且后续治疗可以在当地医院完成。
(二)从基层医院转诊到上级医院
出现以下情况可从基层医院向上级医院转诊。转诊前需严格评估病情,必要时需输血支持治疗。转诊过程中需严密监测生命体征,需备有必要的急救设施及药品。
1. 诊断及疾病状态不明确,需要进一步特殊检查;
2. 基层医院无法处理的严重或进行性溶血状态;
3. 当前的诊断、治疗和随诊无法在基层医院完成。
十、附表
表1 AIHA的直接抗人球蛋白试验种类

表2 儿童原发性AIHA 实验室、病理生理和治疗特点

北京市朝阳区潘家园南里19号人卫大厦
© 2021 人民卫生出版集团 北京人卫智数科技有限公司 京ICP备:19054460号 京公网安备 11010502039674号
京公网安备 11010502039674号

下载APP

公众号

营业执照

出版物经营许可证

 收藏
收藏 已收藏
已收藏



