


 收藏
收藏 已收藏
已收藏磷酸酶症包括低磷酸酶症(hypophosphatasia)和高磷酸酶症(hyperphosphatasia)两种。前者的病因和发病机制较清楚,主要与组织非特异性碱性磷酸酶(tissue-nonspecific alkaline phosphatase,TNAP)基因突变有关;后者的病因未明,TNAP基因无异常,可能与OPG/RANK的结构和功能异常有关,血ALP升高只是一种继发性表现,故高磷酸酶症可能不属于磷酸酶症范畴。
骨组织的酶对骨代谢的调节作用较其他组织明显,能调节骨代谢的酶类很多。本节主要介绍碱性磷酸酶,酸性磷酸酶和骨基质金属蛋白酶-组织骨基质金属蛋白酶-抑制因子系统。
低磷酸酶症(hypophosphatasia)的病因为ALPL突变和血清碱性磷酸酶活性显著降低[5,6]。临床特征是骨骼牙齿矿化缺陷和血清与骨骼的碱性磷酸酶(alkaline phosphatase;OMIM 146300,241500/241510)活性低下,发病率约为1/100 000。低磷酸酶症的临床表现(表1)的变异度极大,严重者出生前死亡,骨骼完全缺乏矿化;轻型患者仅有牙齿脱落而缺乏骨骼病变。临床常分为六种类型,即产前致命型(perinatal lethal type)、产前良性型(perinatal benign type)、幼儿型(infantile type)、儿童型(childhood type)、成年型(adult type)和牙型低磷酸酶症(odontohypophosphatasia type),见表 2。
表1 低磷酸酶症的临床分类
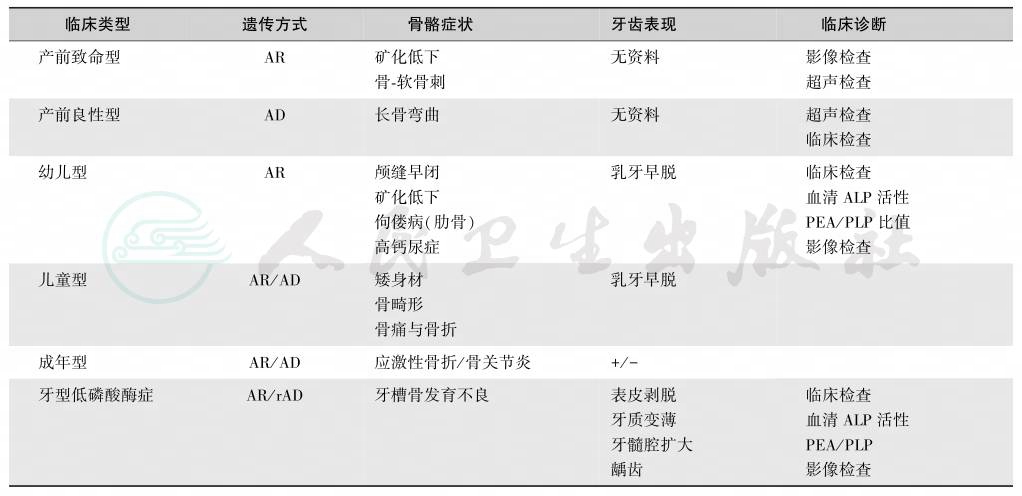
注:PLP:吡多醛-5’-膦酸;PEA:膦酸乙醇胺;AR:常染色体隐性遗传;AD:常染色体显性遗传
表2 ALPL基因突变类型

1型突变引起的后果严重,ALP蛋白积聚于细胞内,并在胞质降解,因而血清和骨骼缺乏ALP活性;2型的ALP也积聚在细胞内,但细胞外和血清中存在一定的ALP活性,由于累及酶活性的关键位点,其临床后果亦相当严重;3型有部分酶在细胞内降解,另一部分可在细胞膜上发挥作用,故体外实验时,酶活性甚至正常或升高,患者的病情较轻。4型的酶活性仅轻度受损,临床症状缺乏或轻微。
低磷酸酶症是一种罕见的代谢性骨病。1948年,由John Campbell Rathbun首先报道。五十多年后,Mumm等从Rathbun报道的婴儿病例的父母的DNA分析中发现,当年他报道的病例的分子病因为TNAP基因两个错义突变(G340A/A881C)。至此,报告的病例达300余例,世界范围内均有发病,所有种族均可受累,通常为常染色体隐性遗传,少数患者有父母近亲婚配史。临床上以骨矿化不足、血清ALP降低以及尿和血液磷酸氨基乙醇(phosphoethanolamine,PEA)含量和血钙升高为特征。多数患者在婴儿或儿童期即可出现症状,如颅骨未骨化或软化,囟门增大并闭合延迟;有些患者因症状轻微可延迟至成人期才被发现。严重者通常在出生后不久即夭折。轻型的儿童病例可仅表现为学步延迟和乳齿早脱。成人患者多因轻微外伤性骨折首次就诊,骨折难以愈合。X线表现为膜化骨和软骨内矿化不足或缺乏。颅骨无矿化,管状骨骨折并畸形,身材矮小和四肢短小畸形。存活的婴幼儿可有不同程度的骨骼受累,主要在骺板和干骺区,其改变与佝偻病相似。成年患者可表现为骨质软化,有时还可见假性骨折(looser带)和少量骨膜下新骨形成。
本病是由于TNAP基因突变所致。至目前为止,已发现突变基因类型80多种。突变类型多为错义突变,少数为复合性突变和无义突变。在欧美,以TNAP基因的E174K突变为最常见;在日本,等位基因的缺失频率高达36%。除假性低磷酸酶症ALP活性正常外,其他类型的低磷酸酶症ALP活性均降低,严重者无法测出。虽然TNAP分布于全身所有的组织中,但TNAP基因突变只引起骨和牙组织病变。Lqbal等分别测定低磷酸酶症的TNAP的免疫活性和生物(催化)活性,得到两者的回归关系式为cBALP(骨源性ALP催化活性)=0.796+3.269 iBALP(骨源性ALP免疫活性)。一般iBALP和cBALP越低,骨骼病变也越严重。如果血清的总TNAP正常,还可测定中性粒细胞 ALP(neutrophil ALP,NAP),得到的计数值(正常人为20~150)如低于15则多为杂合子突变患者。同样,测定ALP同工酶也有相当价值,凡有骨骼病变者,骨源性 ALP将显著降低。另一方面,肝型ALP降低者,可无临床表现。
TNAP基因突变分为三种类型:①第一类突变:如R54C、R54P、A94T、R206W、G317D 和 V365I使 TNAP活性完全丧失,于宫内或出生后不久夭折。②第二类突变:如 A16V、A115V、A160T、A162T、E174K、E174G、D277A、E281K、D361V和G439R使TNAP的活性部分丧失,但严重程度可有较大差别,有些突变仅使酶对一个底物的催化活性丧失,可仅有牙组织的TNAP活性下降,而对另一底物的催化活性完全正常。如A160T突变时,催化对-硝苯磷酸盐(p-nitrophenylphosphate,PNPP)的活性下降,而对PPi的催化活性正常。而D227A突变后的活性变化与A160T刚好相反。③第三类突变:如E174G、E174K、E281K使酶对 PNPP和PPi的催化活性均正常,但对PLP无催化活性,使PLP在脑组织中堆积中毒,导致癫痫样发作。
TNAP突变主要发生于分子链的五个关键区段,即活性位点(active site)及其邻近活性位点凹陷区(active site valley)、同二聚体界面(homodimer interface)、冠状结构域(crown domain)和金属离子(Ca2+)结合位点(mental-binding site),其中后两种结构域为哺乳动物胎盘ALP(placental ALP)所特有。冠状结构域含有胶原结合环(collagen binding loop)。一般引起酶活性显著下降的突变多位于Ca2+结合位点及其附近。临床症状取决于突变部位及程度。成骨细胞分泌的ALP和浆细胞膜型糖蛋白-1是两种相互制约的调节因子,TNAP活性不足,堆积的PPi抑制羟磷灰石结晶的形成和生长。浆细胞膜型糖蛋白-1(plasma cell membrane glycoprotein-1,PC-1)在膜限制性基质囊泡(membrane-limited matrix vesicle,MV)的内侧面表达,PC-1基因突变小鼠表现为矿化过度,导致骨关节炎和脊椎后纵韧带钙化,而 PC-1基因和TNAP基因均缺失的小鼠又表现为正常的矿化功能。因此,凡能抑制PC-1功能的举措都有可能成为治疗低磷酸酶症的途径。
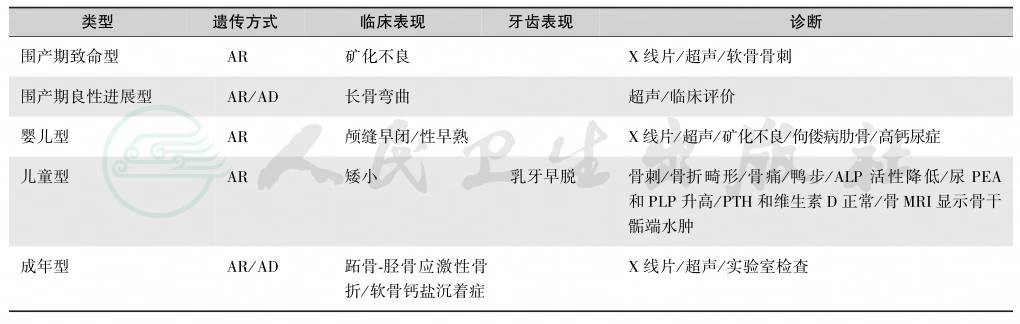
见表3。
表3 低磷酸酶症的临床特点
1.围产型
均为死产或产后不久死亡,表现为骨矿化不良,四肢短,先天性矮小,有的死产儿几乎无骨的矿化。
2.婴幼儿型
有生长障碍,常因抵抗力低而并发肺炎,并常出现高钙血症和高钙尿症,身材矮小,呈佝偻病样畸形;严重病例常伴频发性维生素B6依赖性惊厥和癫痫样发作,维生素B6补充可终止发作。
3.儿童型
出牙迟,牙齿发育不良,儿童型低磷酸酶症(childhood-type hypophosphatasia)的表型多变,乳牙过早脱落为其常见和特征性表现。肢体短(低于正常的2 SD),肌张力低下;前额增大,全身骨的矿化延迟落后,伴肾石病或肾钙盐沉着症,BMD降低。全身生长发育不良,体重低,有些患者可伴有颅缝早闭(craniosynostosis)。TNAP活性可降至正常的5%以下。学步晚,青春期发育延迟。
4.成年型
软骨发育不良,反复发生应激性骨折,造成肢体畸形(图1和图2),中年以后发病者少有佝偻病病史。
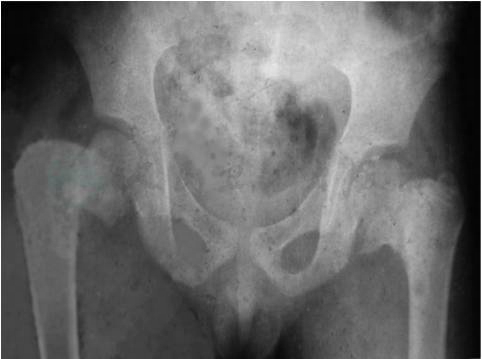
图1 低磷酸酶症(骨盆)
男,6岁,低磷酸酶症骨盆正位片显示骨盆广泛骨密度降低,干骺端变形,颈干角变小
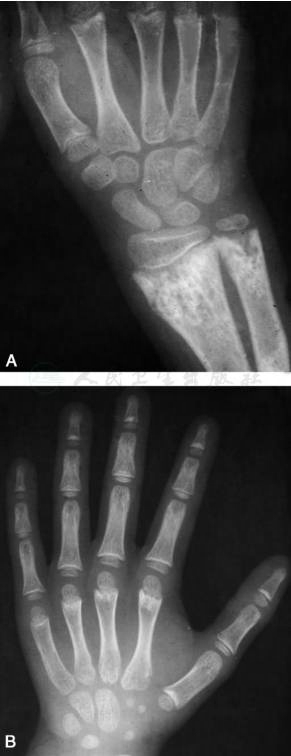
图2 低磷酸酶症(腕部与手部)
女,11岁,A.右腕正位片显示诸骨有轻度骨密度降低,第2、3、4掌骨,尺、桡骨远侧,第1掌骨近侧干骺端呈杯口状凹陷,边缘外展,有粗糙骨小梁和不规则密度减低区,以尺骨较重;B.手部诸骨有轻度骨密度降低,第1掌骨近侧,第2、3、4掌骨远侧及远节指骨近侧干骺端呈杯口状凹陷、有粗糙骨小梁和不规则密度减低区
5.牙型
仅有牙齿的表现。由于牙冠矿化受阻,使牙槽骨间隙小,牙齿生长异常。Watanabe等报道,TNAP的杂合子突变(第9号外显子的T1155C突变来自母方而第10号外显子的G1320A突变来自父方)表现为牙型低磷酸酶症,伴严重牙周炎和牙病变。牙槽和牙发育不良,除乳牙早脱外,还可伴有牙根吸收、牙釉质发育不良、颌骨异常。在光镜、偏振光显微镜和扫描电镜下,可见牙根有大量的骨吸收灶,尤以底部突出,牙本质矿化不良,牙骨质(root cementum)发育不良或无发育,根部牙本质亦有吸收(图3)[7-11]。
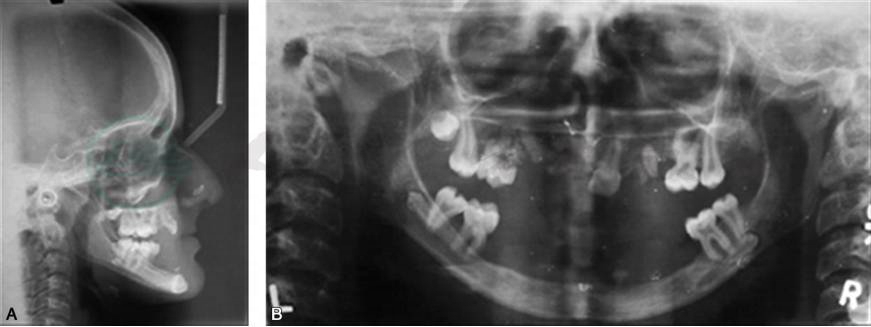
图3 低磷酸酶症的牙齿病变
A.右侧位照片见下颌骨前突和牙齿缺失与发育不良;B.牙槽嵴明显萎缩伴有部分先天性无牙
6.假性低磷酸酶症
与上述儿童型、成人型的临床表现及X线表现相同,但血ALP正常。一些观察结果提示,上述的分类方法可能过于粗糙,各型中还包括不少的亚型。例如在围产型中,Pauli和Moore等报道5例患者有严重骨发育不良,酷似重型成骨不全。但出生后的病程呈良性过程,有长骨发育,其他病变亦有自发缓解趋势,称为良性胎儿型低磷酸酶症(benign prenatal form of hypophosphatasia)。
本病具有部分自限性,有些患者可自愈。病情较重者应采用综合性治疗。
临床上遇到牙齿脱落早、低骨量和骨质增生时,需进行血ALP检测,必要时行ALPL基因筛查;该病可以出现高钙血症,慎用钙剂和维生素D制剂,以防出现肾脏副作用;由于该病可引起骨矿化障碍,使用二膦酸盐会抑制骨吸收,加重骨矿化障碍,慎用二膦酸盐[30,31]。成年型患者发生应激性骨折时,特立帕肽(PTH1-34)可改善预后,但效果不佳。酶替代治疗和骨髓细胞移植为重症患者提供了ALP活性正常的成骨细胞,已经用于本病的治疗[32,33]。二膦酸盐治疗有一定效果,但长期使用可引起非典型骨折[34,35]。ENB-0040(asfotase alfa,Enobia Pharma)是一种重组的融合蛋白,分子中含有TNSALP的细胞外结构域、人IgG1的Fc结构域,分子末端为具有骨骼靶向作用的10肽天冬酰胺。首次静脉注射2mg/kg,继而每周皮下注射3次(1mg/kg);如果效果不佳,可增加剂量至3mg/kg,治疗总疗程可达3年。据报道,该种酶制剂能显著改善病情[36-38]。
1.Chung TD,Sergienko E,Millán JL.Assay format as a critical success factor for identification of novel inhibitor chemotypes of tissue-nonspecific alkaline phosphatase from high-throughput screening.Molecules,2010,15(5):3010-3037.
2.Kawada A,Kashima A,Shiraishi H,et al.Pyridoxine-induced photosensitivity and hypophosphatasia.Dermatology,2000,201(4):356-360.
3.Watanabe H,Goseki-Sone M,Orimo H,et al.Function of mutant(G1144A)tissue-nonspecific ALP gene from hypophosphatasia.J Bone Miner Res,2002,17(11):1945-1948.
4.Mornet E.Hypophosphatasia.Orphanet J Rare Dis,2007,2:40.
5.Zhang H,Ke YH,Wang C,et al.Identification of the mutations in the tissue-nonspecific alkaline phosphatase gene in two Chinese families with hypophosphatasia.Arch Med Res,2012,43(1):21-30.
6.Satou Y,Al-Shawafi HA,Sultana S,et al.Disulfide bonds are critical for tissue-nonspecific alkaline phosphatase function revealed by analysis of mutant proteins bearing a C(201)-Y or C(489)-S substitution associated with severe hypophosphatasia.Biochim Biophys Acta,2012,1822(4):581-588.
7.秦满,矿香,曷立宏.低磷酸酯酶症乳牙的光镜和扫描电镜研究.中华口腔医学杂志,1999,34(4):220-222.
8.Chappie IL.Hypophosphatasia.Dental aspects and mode of inheritance.J Clin Periodontol,1993,20:615-622.
9.Fraser D.Hypophosphatasia.Am J Med,1957,22:730-746.
10.Beumer J,Trowbridge HO,Silverman S,et al.Childhood hypophosphatasia and the premature loss of teeth.A clinical and laboratory study of seven cases.Oral Surg Oral Med Oral Pathol,1973,35:631-640.
11.Bagis B,Baltacioglu E,Aydogan E,et al.Prosthetic rehabilitation of hypophosphatasia:a case report.Cases J,2008,12:7626.
12.Taketani T.Neurological Symptoms of Hypophosphatasia.Subcell Biochem,2015,76:309-322.
13.Whyte MP,Greenberg CR,Salman NJ,et al.Enzyme-replacement therapy in life-threatening hypophosphatasia.N Engl J Med,2012,366(10):904-913.
14.Orimo H,Hayashi Z,Watanabe A,et al.Novel missense and frameshift mutations in the tissue-nonspecific alkaline phosphatase gene in a Japanese patient with hypophosphatasia.Hum Mol Genet,1994,3:1683-1684.
15.Orimo H,Goseki-Sone M,Sato S,et al.Detection of deletion 1154-1156 hypophosphatasia mutation using TNSALP exon amplification.Genomics,1997,42:364-366.
16.Mornet E,Taillandier A,Peyramaure S,et al.Identification of fifteen novel mutationsin the tissue-nonspecific alkaline phosphatase(TNSALP)gene in European patients with severe hypophosphatasia.Eur J Hum Genet,1998,6:308-314.
17.Goseki-Sone M,Orimo H,Iimura T,et al.Hypophosphatasia:identification of five novel missense mutations(G507A,G705A,A748G,T1155C,G1320A)in the tissue-nonspecific alkaline phosphatase gene among Japanese patients.Hum Mutat,1998,S263-S267.
18.Mumm S,Jones J,Finnegan P,et al.Denaturing gradient gel electrophoresis analysis of the tissue nonspecific alkaline phosphatase isoenzyme gene in hypophosphatasia.Mol Genet Metab,2002,75:143-153.
19.Watanabe H,Hashimoto-Uoshima M,Goseki-Sone M,et al.A novel point mutation(C571T)in the tissue-non-specific alkaline phosphatase gene in a case of adult-type hypophosphatasia.Oral Dis,2001,7:331-335.
20.Watanabe H,Goseki-Sone M,Iimura T,et al.Molecular diagnosis of hypophosphatasia with severe periodontitis.J Periodontol,1999,70:688-691.
21.Whyte MP,Mumm S,Deal C.Adult hypophosphatasia treated with teriparatide.J Clin Endocrinol Metab,2007,92:1203-1208.
22.Watanabe A,Yamamasu S,Shinagawa T,et al.Prenatal genetic diagnosis of severe perinatal( lethal) hypophosphatasia.J Nippon Med Sch,2007,74:65-69.
23.Henthorn PS,Whyte MP.Infantile hypophosphatasia:successful prenatal assessment by testing for tissue-non-specific alkaline phosphatase isoenzyme gene mutations.Prenat Diagn,1995,15:1001-1006.
24.Orimo H,Nakajima E,Hayashi Z,et al.First-trimester prenatal molecular diagnosis of infantile hypophosphatasia in a Japanese family.Prenat Diagn,1996,16:559-563.
25.Mornet E,Muller F,Ngo S,et al.Correlation of alkaline phosphatase(ALP)determination and analysis of the tissue non-specific ALP gene in prenatal diagnosis of severe hypophosphatasia.Prenat Diagn,1999,19:755-757.
26.Girschick HJ,Schneider P,Kruse K,et al.Bone metabolism and bone mineral density in childhood hypophosphatasia.Bone,1999,25(3):361-367.
27.Vandevijver N,De Die-Smulders CE,Offermans JP,et al.Lethal hypophosphatasia,spur type:case report and fetopathological study.Genet Couns,1998,9(3):205-209.
28.Shibata H,Fukushi M,Igarashi A,et al.Defective intracellular transport of tissue-nonspecific alkaline phosphatase with an Ala 162→Thr mutation associated with lethal hypophosphatasia.J Biochem(Tokyo,Abstract),1998,123(5):968-977.
29.Krawitz PM,Schweiger MR,Rödelsperger C,et al.Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome.Nat Genet,2010,42(10):827-829.
30.Wenkert D,Podgornik MN,Coburn SP,et al.Dietary phosphate restriction therapy for hypophosphatasia:preliminaryobservations.Fifth International Alkaline Phosphatase Symposium:“Understanding alkaline phosphatase function-Pathophysiology and treatment of Hypophosphatasia and other AP-related diseases” 2007,Huningue,France.2007.
31.Girschick HJ,Seyberth HW,Huppertz HI.Treatment of childhood hypophosphatasia with nonsteroidal antiinflammatory drugs.Bone,1999,25:603-607.
32.Laroche M.Failure of teriparatide in treatment of bone complications of adult hypophosphatasia.Calcif Tissue Int,2012,90(3):250.
33.Cahill RA,Wenkert D,Perlman SA,et al.Infantile Hypophosphatasia:Transplantation Therapy Trial Using Bone Fragments and Cultured Osteoblasts.J Clin Endocrinol Metab,2007,92(8):2923-2930.
34.Whyte MP,Greenberg CR,Salman NJ,et al.Enzyme-replacement therapy in life-threatening hypophosphatasia.N Engl J Med,2012,366(10):904-913.
35.Sutton RA,Mumm S,Coburn SP,et al.“Atypical femoral fractures”during bisphosphonate exposure in adult hypophosphatasia.J Bone Miner Res,2012,27(5):987-994.
36.Whyte MP,Greenberg CR,Salman NJ,et al.Enzyme-replacement therapy in life-threatening hypophosphatasia.N Engl J Med,2012,366(10):904-913.
37.Hofmann C,Jakob F,Seefried L,et al.Recombinant enzyme replacement therapy in hypophosphatasia.Subcell Biochem,2015,76:323-341.
38.Liu J,Campbell C,Nam HK,et al.Enzyme replacement for craniofacial skeletal defects and craniosynostosis in murine hypophosphatasia.Bone,2015,78:203-211.









