


 收藏
收藏 已收藏
已收藏英文名称 :Phosphatase disease
磷酸酶病包括低磷酸酶症(hypophosphatasia)和高磷酸酶症(hyperphosphatasia)两种。前者的病因和发病机制较清楚,主要与组织非特异性碱性磷酸酶(tissue-nonspecific alkaline phosphatase,TNAP)基因突变有关;后者的病因未明,TNAP基因无异常,可能与OPG/RANK的结构和功能异常有关,血ALP升高只是一种继发性表现,故高磷酸酶症可能不属于磷酸酶病范畴。
1.低磷酸酶症
本病是由于TNAP基因突变所致。至目前为止,已发现突变基因类型八十多种。突变类型多为错义突变,少数为复合性突变和无义突变。在欧美,以TNAP基因的E174K突变为最常见;在日本,等位基因的缺失频率高达36%。
除假性低磷酸酶症ALP活性正常外,其他类型的低磷酸酶症ALP活性均降低,严重者无法测出。虽然TNAP分布于全身所有的组织中,但TNAP基因突变只引起骨和牙组织病变。Iqbal等分别测定低磷酸酶症的TNAP的免疫活性和生物(催化)活性,得到两者的回归关系式为cBALP(骨源性ALP催化活性)=0.796+3.269iBALP(骨源性ALP免疫活性)。一般iBALP和cBALP越低,骨骼病变也越严重。如果血清的总TNAP正常,还可测定中性粒细胞ALP(neutrophil ALP,NAP),得到的计数值(正常人为20~150)如低于15则多为杂合子突变患者。同样,测定ALP同工酶也有相当价值,凡有骨骼病变者,骨源性ALP将显著降低。另一方面,肝型ALP降低者,可无临床表现。
(1)第一类突变
如R54C、R54P、A94T、R206W、G317D和V365I使TNAP活性完全丧失,于宫内或出生后不久夭折。
(2)第二类突变
如A16V、A115V、A160T、A162T、E174K、E174G、D277A、E281K、D361V和G439R使TNAP的活性部分丧失,但严重程度可有较大差别,有些突变仅使酶对一个底物的催化活性丧失,可仅有牙组织的TNAP活性下降,而对另一底物的催化活性完全正常。如A160T突变时,催化对-硝苯磷酸盐(p-nitrophenylphosphate,PNPP)的活性下降,而对PPi的催化活性正常。而D227A突变后的活性变化与A160T刚好相反。
(3)第三类突变
如E174G、E174K、E281K使酶对PNPP和PPi的催化活性均正常,但对PLP无催化活性,使PLP在脑组织中堆积中毒,导致癫痫样发作。
TNAP突变主要发生于分子链的5个关键区段,即活性位点(active site)及其邻近活性位点凹陷区(active site valley)、同二聚体界面(bomodimer interface)、冠状结构域(crown domain)和金属离子(Ca2+)结合位点(mental-binding site),其中后两种结构域为哺乳动物胎盘ALP(placental ALP)所特有。冠状结构域含有胶原结合环(collagen binding loop)。一般引起酶活性显著下降的突变多位于Ca2+结合位点及其附近。临床症状取决于突变部位及程度。
成骨细胞分泌的ALP和浆细胞膜型糖蛋白-1是两种相互制约的调节因子,TNAP活性不足,堆积的PPi抑制羟磷灰石结晶的形成和生长。浆细胞膜型糖蛋白-1(plasma cell membrane glycoprotein-1,PC-1)仅存在于膜限制性基质囊泡(membrane-limited matrix vesicles,MVs)的内侧面,PC-1基因突变小鼠表现为矿化过度,导致骨关节炎和脊椎后纵韧带钙化,而PC-1基因和TNAP基因均缺失的小鼠又表现为正常的矿化功能。因此,凡能抑制PC-1功能的举措都有可能成为治疗低磷酸酶症的途径。
2.高磷酸酶症
又称遗传性高磷酸酶症(hereditary hyperphosphatasia)、幼年Paget骨病(juvenile Paget disease)、慢性高磷酸酶骨病(chronic osteopathy with hyperphosphatasia)或高磷酸酶性骨膨大症。本病首先由Bakwin和Elger(1956年)报道,目前已有数十例报道,患儿因骨构塑异常而导致严重骨畸形。其特点是血清特发性ALP增高。血清ALP增高的原因未明。遗传性高磷酸酶症伴精神发育迟钝综合征(hyperphosphatasia mental retardation,HPMR syndrome)为常染色体隐性遗传性疾病,患者的精神障碍和面部有特征性表现,血清碱性磷酸酶明显升高,目前证明,至少与编码GPI-锚的生物合成途径的因子(a member of the GPI-anchor biosynthesis pathway)突变相关。
骨形成和骨吸收均显著增强,代谢转换率明显升高。Cundy认为与OPG功能异常(OPG基因失活性突变)有关。受累者身材矮小,头大,管状骨呈弥漫性肥厚弯曲而易骨折,患者活动严重受限。本病为常染色体隐性或显性遗传,表型不均一,即使在同一家庭,受累程度也不一致,好发于婴幼儿和儿童期,患者多早年夭折。至此,文献所报道的患者年龄最大者为38岁。病理检查发现骨细胞所产生的骨基质和胶原均过多,原始交织骨不能转化为成熟的板层骨,同时伴有皮质骨(致密骨)的分层钙化及皮肤色素沉着。
1.碱性磷酸酶及其同工酶与骨代谢关系密切
ALP广泛存在于肝、骨骼、肾、小肠及胎盘等组织中,不同组织中的ALP功能不同;且同一组织的ALP同工酶在不同细胞和亚细胞结构中也有不同的作用。ALP的同工酶有4种,其中肝、骨骼、肾脏中的ALP称为组织非特异性碱性磷酸酶(tissue-nonspecific alkaline phosphatase,TNAP)。
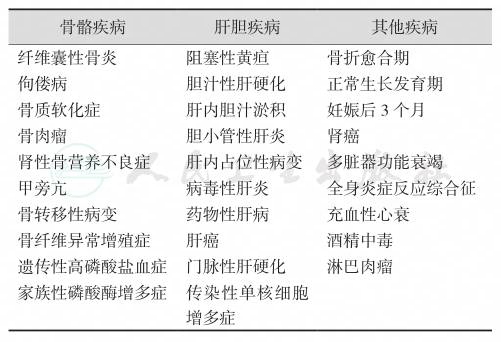
由于除骨以外的其他组织也能合成和释放ALP,所以除骨组织本身的疾病外,其他如肝、肠、肾脏的疾病和某些药物均可引起ALP活性增高(表1)或降低(如低磷血症、甲状旁腺功能减退症、家族性磷酸酶过少症等)。同时,除疾病以外,ALP还受年龄和性别的影响。儿童血清中有大量的骨源性ALP,而极少或甚至没有肝和肠ALP。50岁以下的成人血清内均有肝、骨和肠来源的ALP。50岁以上者,血清骨源性ALP活性升高。
表1 引起血清ALP活性升高的主要疾病
2.骨源性ALP是骨形成标志物
在基质囊泡(matrix vesicle)内可发现活性很高的ALP,基质小泡是骨矿化中心,因此ALP与骨矿化有密切关系。此外,用婴幼儿低磷酸酶症的TNAP基因敲除模型发现,TNAP还可双相性调节成骨细胞的浆细胞膜型糖蛋白-1(PC-1)和磷酸二酯酶-核苷焦磷酸酶(phosphodiesterase nucleotide pyrophosphatase,PDNP/NTPPPH)。ALP在碱性(pH 8~10)条件下,催化磷酸单脂分解,形成的非特异性水解酶催化无机磷酸盐水解,降低焦磷酸盐浓度,有利于骨矿化(无机焦磷酸盐是矿化作用的极强抑制剂)。
骨化中心矿化时,在骨干软骨膜和肥大型软骨细胞处形成一薄层类骨质,围绕着骨干扩展,形成环状或领口样类骨质层,此时已存在ALP活性。这些细胞与胶原纤维排列成束,形成骨小梁。软骨中的ALP来源于肥大型软骨细胞。基质的矿化有时与原始骨同时形成,但多在其后出现。当板层骨沉积活跃时,骨表面被成骨细胞覆盖,其胞质内含有核糖核酸和较多的线粒体,使成骨细胞胞质呈碱性。分化了的成骨细胞含有糖原,一旦这些细胞形成骨组织,糖原即消失。与之相邻的成骨细胞和组织也含有ALP,但当骨矿化开始时,ALP活性即开始下降。因此,无论胚胎期的骨形成过程中或在成人,成骨细胞在数量和(或)功能增加、降低时,ALP的活性与之同步消长。
临床上很早就应用血清ALP活性测定来诊断骨骼疾病。骨病的诊断可借助X线、闪烁扫描、磁共振等来诊断,ALP在一些骨病的早期改变与形态学变化一致,所以ALP活性测定对骨病的早期诊断和鉴别诊断有重要价值,而且是提供病情随访和预后的重要依据。
1.低磷酸酶症
本病呈部分自限性,有些患者可自愈。病情较重者应采用综合性治疗。曾有学者采用静脉滴注几种具活性的碱性磷酸酶治疗,虽有生物化学上的改善(如血钙、磷、ALP基本正常),但随着酶在体内降解,活性消退,半年观察未见有放射学的改变。降钙素和氯噻嗪可纠正高钙血症,抑制骨钙释放和细胞外液钙转移。常用的有益盖宁或密钙息,用量根据病情及血钙水平确定。氯噻嗪能减轻高钙尿和骨的低矿化,从而间接降低高血钙。常用量为每次25mg,25~75mg/d。非甾体类固醇性消炎止痛药有助于缓解患者的疼痛(抑制前列腺素E的合成和释放)。因制动或肌无力,患者少活动而使机体抵抗力降低,特别是婴幼儿或严重病例有50%以上易并发肺部感染而死亡,故应补充足够的蛋白质、糖类及微量元素和维生素,以满足生长发育的需要,个别行走不便的患儿应注意被动按摩和关节的屈曲运动,同时加强看护,以免碰撞跌倒造成骨折。在动物实验中,维生素B6可治疗TNAP基因敲除(-/-)小鼠的癫痫样发作。同样,正常小鼠如严重缺失维生素B6,又可诱发如同TNAP基因敲除鼠相似的癫痫发作,但骨的代谢与成骨细胞功能不受影响。故可认为骨骼组织外的许多病变与维生素B6代谢失常有关,而骨病变与维生素B6代谢异常无关。维生素B6可诱导本病患者(如TNAP基因1154-1156杂合缺失)的紫外线过敏,其原因可能与维生素B6代谢异常有关。
2.高磷酸酶症
近年应用羟乙磷酸钠或帕米磷酸钠治疗取得了一定疗效。使用时间在5周以上,血ALP可下降40%~70%,尿羟脯氨酸可下降至正常水平,而无明显的不良反应。降钙素除对于骨折造成的骨痛有明显止痛作用外,还可改善骨病变,缓解病情。









