


 收藏
收藏 已收藏
已收藏英文名称 :spondyloepiphyseal dysplasia
脊柱-骨骺发育不良症(spondyloepiphyseal dysplasia,SED)指主要累及脊柱骨骺和长骨末端的进行性骨软骨发育不良,其特征表现为短躯干性侏儒和继发性骨关节炎。
脊柱-骨骺发育不良症分为先天性(congenita SEDL)和迟发性(tarda SEDT)两类。此外,还包括假性软骨发育不良(pseudoachondroplasia);晚发型脊柱-骨骺发育不良伴进行性骨关节病(spondyloepiphyseal dysplasia tarda with progressive arthropathy,SEDT-PA)于1982年首先由Wynne-Davies等报道,属脊柱-骨骺发育不良症的一种特殊类型。
目前所知,上述先天性和迟发性脊柱-骨骺发育不良症、假性软骨发育不良及晚发型脊柱-骨骺发育不良伴进行性骨关节病都属于遗传性疾病。
(一)COL2A1突变导致先天性脊柱骨骺发育不良症
先天性脊柱-骨骺发育不良症(spondyloepiphyseal dysplasia congenita,SDC,OMIM183900)为常染色体显性遗传骨发育性疾病,致病基因COL2A1定位于12q13.11-q13.2。已知突变类型有:Gly154Arg、Arg519Cys、Arg789Cys、Gly997Ser、Gly973Arg、Thr1370Met、1-exon Del(1号外显子缺失)和45-bp Dup Ex48。
(二)迟发性脊柱-骨骺发育不良症包括三种遗传方式
迟发性脊柱-骨骺发育不良症(spondyloepiphyseal dysplasia tarda,SEDT)患儿出生时无任何异常,儿童期发病,属单基因遗传病,具有高度遗传异质性,符合孟德尔遗传规律。Carter和Sutcliffe于1970年指出,迟发性脊柱-骨骺发育不良症共有常染色体显性遗传、常染色体隐性遗传和X-连锁隐性遗传3种遗传方式。由于基因型不同,表型也有各自的特点。因此,严格来说SEDT包括了3种不同的疾病。
(三)SEDLIN突变引起X-连锁型SEDT
X-连锁型SEDT又称晚发性脊柱-骨骺发育不良(spondyloepiphyseal dysplasia late,SEDL,OMIM313400),属性染色体连锁遗传病,呈X-连锁隐性遗传,仅男性发病。致病基因为SEDLIN,定位于Xp22.2-p22.1,含有6个外显子。已知突变类型包括缺失突变、插入突变和置换突变3种类型。已有文献报道的缺失突变分别有:3号外显子的53和54位缺失两个T(2-bp Del,53TT)、3号外显子的157和158位缺失AT(2-bp Del,157AT)、4号外显子的191和192位缺失TG(2-bp Del,191TG)、5号外显子的267~271位缺失AAGAC(5-bp Del,NT267)、5号外显子的271~275位缺失(5-bp Del,NT271)和6号外显子的613位缺失A(1-bp Del,613A)共六种,分别导致移码突变和截短蛋白。插入突变有:3号内含子剪接区的插入突变(IVS3+5G-A)和4号内含子与5号外显子处的插入突变(IVS4+4T-C)两种。置换突变有:F83S、L131T和S110T共3种。
(四)AGC1突变导致常染色体显性遗传型SEDT
常染色体显性遗传型SEDT(OMIM608361)的致病基因可能为AGGRECAN(AGC1),定位于15q26.1。已知突变类型有12外显子插入1个(1-bp INS),导致aggrecan蛋白在212个氨基酸残基处出现移码突变并形成截短蛋白,AGC1基因突变可能导致疾病的发生,是引起脊柱-骨骺发育不良症的突变基因谱中的一个致病基因。常染色体隐性遗传型SEDT的致病基因尚不清楚,Leroy等于2004年报道2个家系是新的SEDT类型为常染色体隐性遗传。
(五)WISP3突变导致晚发型脊柱-骨骺发育不良伴进行性骨关节病
本病又称儿童进行性假类风湿性关节病(arthropathy progressive pseudo-rheumatoid of childhood,APPRC)或 进 行性假类风湿性骨发育不良(progressive pseudorheumatoid dysplasia,PPD,OMIM208230),属常染色体隐性遗传病,男女发病率类似。致病基因为WISP3,定位于6q22-q23,编码含354个氨基酸残基且富含半胱氨酸的分泌型蛋白,相对分子量约40kD。已知突变类型有:(1-bpDel246A)、IVS1DS、1-bpINS+2T、2-bpINS863AC)、2-bpDel43GC、2-bpDel866AG、1-bpDel840T、S334P、C145Y、W331T、C52T和C78R,其中C78R也可能为单核苷酸基因多态性。WISP3蛋白由4个结构域构成。结构域1与胰岛素样生长因子结合蛋白(IGFBPs)1-6富含半胱氨酸的N-末端具有约32%的同源性;结构域2包含一个von Willebrand型C结构域(VWC),这种结构域存在于von Willebrand因子以及不同种类的黏蛋白、血小板反应蛋白和胶原纤维中,因此,结构域2可能参与CCN家族蛋白寡聚化过程;结构域3是血小板反应Ⅰ型蛋白,含有一个WSXCSXXCG位点,是一个细胞接触位点,它可结合硫酸甘氨酸结合物;结构域4是C-末端模块,含有10个半胱氨酸残基,其中的6个半胱氨酸集合成胱氨酸花结,这种花结也出现在神经生长因子(NCF)、转化生长因子β(TGF-β)和血小板性生长因子(PDGF)中,C-末端模块可能含有二聚化和受体结合区域,参与了细胞表面受体的结合。
WISP3蛋白产物的完整性是维持正常软骨代谢的关键因素。最近对SEDT-PA基因突变的功能表达研究表明CCN6基因可能通过调节Ⅱ型胶原和aggrecan在软骨基质中的表达,维持软骨的完整性,而SEDT-PA相关基因突变可引起这种功能的丧失,导致持续性软骨丢失,可能部分解释SEDT-PA的发病机制。
(六)假性软骨发育不良具有遗传异质性
其中Ⅰ、Ⅲ型为常染色体显性遗传方式,Ⅱ、Ⅳ型为常染色体隐性遗传方式,亦可为散发。Maroteaux等认为本病是脊柱骨骺发育不良的一个特殊类型(肢体型),具体发病机制尚待进一步研究。
近年报道,COMP 基因可能是假性软骨发育不良的候选致病基因,定位于19p13.1。Comp蛋白质分子量约524kD,在软骨基质中高水平表达,软骨基质中Ⅰ、Ⅱ和Ⅸ胶原均与Comp蛋白有高结合亲和力。COMP 基因突变致假性软骨发育不良的突变位点有:D472Y、C468Y、C328R、D473G、G719D、C348Arg、3-bpDel1375-1377TCA、Ser459Del、3-bpDelAsp、Del(GAC)4、(GAC)5、(GAC)7和EX9Del,其中D472Y和D473G也可能为单核苷酸基因多态性。
脊柱-骨骺发育不良症是一组进行性不可逆的疾病,目前尚无有效的治疗办法,应以预防为主。由于尚未发现生化代谢异常,故不能用生化方法进行产前诊断。
可根据病史包括疾病家族史、患者症状、体征及X线检查明确其临床诊断,然后行致病基因突变检测,明确致病基因、突变位点及遗传方式,针对其开展遗传咨询及胎儿产前诊断,以防止遗传病的下传,最终达到优生的目的。例如比较简便的做法可根据X-连锁晚发性脊柱骨骺发育不良症的性染色体隐性遗传规律,通过产前预测胎儿性别,舍去男胎,达到防止患病个体出生的目的。
针对脊柱-骨骺发育不良这一组疾病的治疗主要是对症治疗,关节过度松弛不稳者可行关节融合术;长骨弯曲畸形者,可截骨矫形,纠正力线;股骨头病变所致的继发性骨关节炎,病变严重影响行走者需行全髋关节置换术。
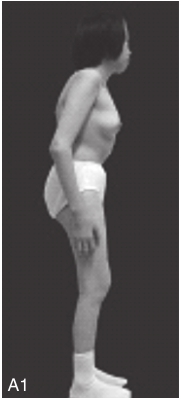


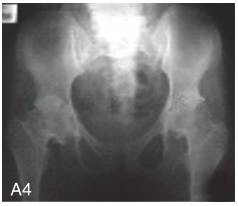
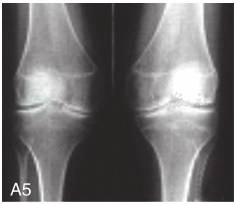
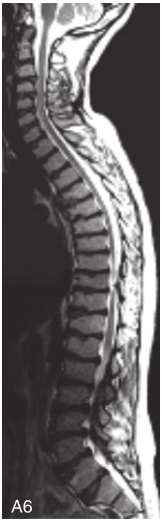
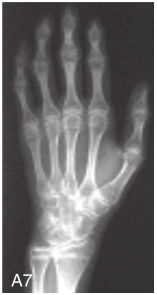
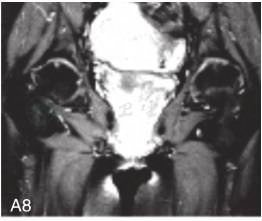
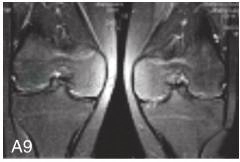
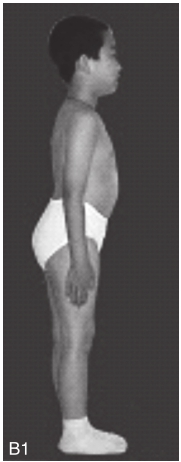
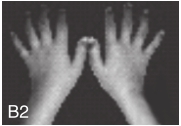
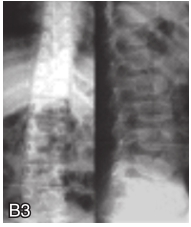
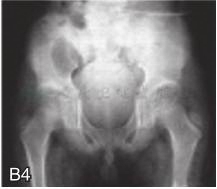

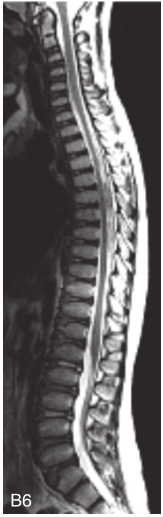
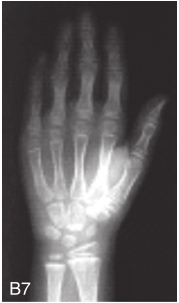
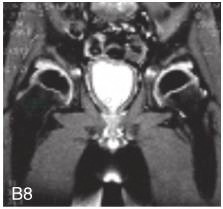
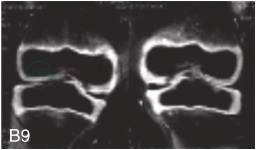
图6-37-92晚发型脊柱骨骺发育不良伴进行性骨关节病
注:一家系中两例患者,为同胞姐弟,均于6岁时开始发病。患者X线表现为椎体普遍变扁而前后径及横径加大,多数椎体上、下缘前部凹陷而中后部稍膨隆或平直,椎间隙变窄,常合并脊柱侧弯,椎弓根前后径变短。髂骨底部宽而短,髂骨翼及骶髂关节相对发育较小。髋关节呈退行性骨关节病改变。四肢管状骨骨质疏松,骨骺和干骺端可对称性增大,并向侧方突出,大关节呈不同程度退行性骨关节病改变,远位指间关节增粗并屈曲,致末节指骨相对变尖和偏斜









