


 收藏
收藏 已收藏
已收藏英文名称 :Paget disease of bone
Paget骨病(Paget disease of bone,PDB)是老年白种人中仅次于骨质疏松的常见代谢性骨病。PDB又称变形性骨炎或畸形性骨炎(osteitis deformans),是局部骨组织的一种骨重建(bone remodeling)亢进性疾病。其病变特点是病灶处的骨重建(骨吸收、骨形成和骨矿化)增加;由于过高的破骨细胞活性及过多的破骨细胞引起高速骨溶解,并导致成骨细胞增加和骨形成过多,形成的交织骨结构脆弱;骨盐及胶原的转换率增加,骨髓纤维化和血管过多致骨局限性膨大。由于骨形成和骨吸收之间失衡导致骨面积增大和骨畸形。Paget骨病可能包括了数种不同的临床类型。
Paget骨病是仅次于骨质疏松的常见代谢性骨病。发病无性别差异,初次就诊年龄多在40岁以上(尚未见有18岁以下的病例报道)。本病具有家族遗传特点,有阳性家族史者一般约占15%,最高达40%。Paget骨病主要流行于盎格鲁撒克逊人(Anglo-Saxon)后代中。据考证,音乐圣师——路德维希·凡·贝多芬(Ludwig van Beethoven,1770~1827年)患的也是Paget骨病。由于过高的破骨细胞活性及过多的破骨细胞引起高速骨溶解,导致成骨细胞增加和骨形成过多,骨髓纤维化和血管增生导致局限性骨膨大和骨畸形,并引起贝多芬失聪。
本病的流行具有显著的地理和人种特异性。例如,当单骨性Paget骨病的印度男性患者迁移到Paget骨病流行区后,其病情明显加重。Paget骨病的高危环境可能主要是病毒感染或其相关因素。本病的地域分布明显,英国的英格兰和威尔士地区最常见,发病率在55岁以上人群中约为0.3%,随着年龄增加,发病率急剧上升。考古分析公元前1850年~公元前900年古英格兰北部居民(2770例)的Paget骨病患病率,经X线照片证实者为15例,患病率为2.1%(40岁以上者)。公元前1500年间为1.7%,而公元后1500年间为3.1%,说明本病在过去的几千年中患病率增加。澳大利亚、新西兰、南非及美国次之,美国Paget骨病的总患病率不低于1%,有的地区超过2%。但近几年有证据表明,英美两国的发病率正在下降,下降的原因可能与环境因素有关。在北欧、中东阿拉伯、中国、日本该病少见。在法国、意大利及西班牙等国,发病率居中。我国和亚洲地区少见,北京、河北、河南、山东、湖南、甘肃及台湾等省均有报道。
(一)前破骨细胞表达病毒组分并诱导破骨细胞生成和骨吸收
1.麻疹病毒感染
1980年,Rebel等在20例Paget骨病患者的破骨细胞中检出麻疹病毒抗原,并在破骨细胞和培养的骨组织细胞中发现麻疹病毒核壳蛋白抗原。1995年,Reddy等发现,在外周血单核细胞中存在麻疹病毒的转录产物。后经人们用克隆麻疹病毒cDNA探针检测,其阳性率达80%~90%,而且除破骨细胞和外周血单核细胞外,在病变部位的成骨细胞、骨细胞、成纤维细胞、单核细胞和淋巴细胞中也存在麻疹病毒感染的证据,但伴有氟骨症、骨折和甲旁亢者为阴性。Kurihara的实验提示,破骨细胞前身细胞表达麻疹病毒核蛋白外壳组分并刺激破骨细胞生成,诱导破骨细胞表达Paget骨病表型的破骨细胞。Niedermeyer等用RT-PCR发现,在83%的耳骨硬化性病变中存在麻疹病毒RNA,淋巴液中的抗麻疹病毒IgG浓度高于血液(女性患者更常见)。因此,提示耳硬化性病变(包括Paget骨病病变)与麻疹病毒感染有密切关系。
2.其他病毒感染
连续病理切片还发现破骨细胞中存在呼吸道融合病毒核壳蛋白抗原。在Paget骨病病变部位的细胞培养和骨切片中发现存在抗呼吸道融合病毒阳性反应,但抗麻疹病毒、副流感病毒、流感病毒A和B、单纯疱疹病毒和风疹病毒的反应均为阴性。一些口腔细菌(如actinobacillus actinomycetemcomitans)具有溶骨作用,分泌的溶骨性物质(62kD)的浓度在pg/L范围内,另一些致牙周炎细菌亦可活化破骨细胞。
超微结构观察发现,Paget骨病的破骨细胞核及细胞质存在杂乱排列的微丝状或束状结晶,以电子密集的细胞核为多,紧密堆积。位于细胞质内的“晶体”多局限于某一部位,有时还存在包涵体(呈条状或梭形结构,有外膜),可能是一种副黏病毒(paramyxovirus)的核蛋白包涵体膜,与麻疹病毒的抗核蛋白外壳抗体有交叉反应。此外,英国的Paget骨病患者多有养狗的嗜好,犬瘟热病毒亦有可能成为致病原。支持病毒学说的基本依据是本病多在40岁以后发病,潜伏期长,发作呈单器官局部亚急性临床过程。本病发作时,骨吸收和骨形成加速,伴纤维变性,为慢性炎症反应;有些病例的发病还具有地区性和家族性特征。病毒感染导致Paget骨病的另一例子是耳骨硬化症(otosclerosis)。
(二)内分泌功能紊乱和自主神经功能障碍促进溶骨
Gutteridge对30例Paget骨病和甲旁亢行甲状旁腺切除术后的跟踪结果支持内分泌功能紊乱假说。肾上腺皮质功能不全患者并无Paget骨病的骨骼异常,相反,本病需要用肾上腺糖皮质激素来缓解疼痛。Paget骨病的发病与患者骨髓基质细胞过度表达RANKL和破骨细胞及其前身细胞对RANKL反应过度敏感有关。用Paget骨病患者的骨髓细胞作培养,破骨细胞前身细胞和破骨细胞可产生大量的IL-6,细胞表达的IL-6受体数量和外周血IL-6亦升高,破骨细胞对1,25-(OH)2D和降钙素的反应性均增强(过敏)、24-羟化酶活性被上调,IL-6再刺激前身细胞转化为成熟的破骨细胞。这说明患者的骨髓微环境中的一些体液因素有利于破骨细胞的生成,并促进其溶骨活性。
滋养骨的动脉血量增加导致骨质局部充血,进而促使成骨细胞代偿性增加和新生骨的异常增加,并可造成骨组织结构紊乱。骨小梁骨化不全,骨皮质为尚未骨化的类骨质替代,皮质和骨髓界限不清,骨质疏松,负重后畸形;继而成骨细胞活性增强,骨质由疏松变脆变硬,容易发生病理性骨折。
病因和发病机制未明,但近年来有重大的新发现。从目前的研究结果看,Paget骨病很可能是一种以局限性高速骨溶解为特征的临床综合征,而高速骨溶解的基本原因是OPGRANK-RANKL信号分子或相关基因的突变。病毒感染、内分泌功能紊乱和自主神经功能紊乱也在Paget 骨病的发病中起了重要作用。因此,Paget骨病是一种基因与环境相互作用而导致疾病的典型例子。
(一)OPG及相关基因突变引起局限性高速骨溶解
本病的家族聚集现象明显,家族性Paget骨病以常染色体显性方式遗传。近年的研究发现,Paget骨病很可能是一种遗传综合征。全基因组扫描发现了4个易感基因或遗传位点。Paget骨病样家族性扩张性溶骨症(PDB-like syndromes of familial expansile osteolysis)、早发性家族性Paget骨病(early-onset familial PDB)和扩张性骨性高磷酸酶血症(expansile skeletal hyperphosphatasia)的分子病因分别为破骨细胞的调节因子RANK插入突变(TNFRSF11A)、护骨素(osteoprotegerin,OPG)失活性突变(TNFRSF11B)和 RANK配体(RANKL)多态性有关(图1)。
Paget骨病致病基因可能还包括SQSTM1以及SQSTM1相关基因。此外,含valosin蛋白(valosin-containing protein,VCP)基因突变除引起Paget骨病外,还导致遗传性包涵体肌病综合征(syndrome of hereditary inclusion body myopathy)和前颞型痴呆(fronto-temporal dementia)。Whyte等发现两例儿童型Paget骨病患者均存在该基因的纯合子缺失(断裂点相同,约缺失100kb)。患者血清中的OPG极低,而可溶性破骨细胞分化因子(sODF)明显升高。因而至少一部分儿童型Paget骨病与TNFRSFⅡB的纯合子缺失导致OPG缺乏有关。青少年Paget骨病常伴有进展性视网膜病。
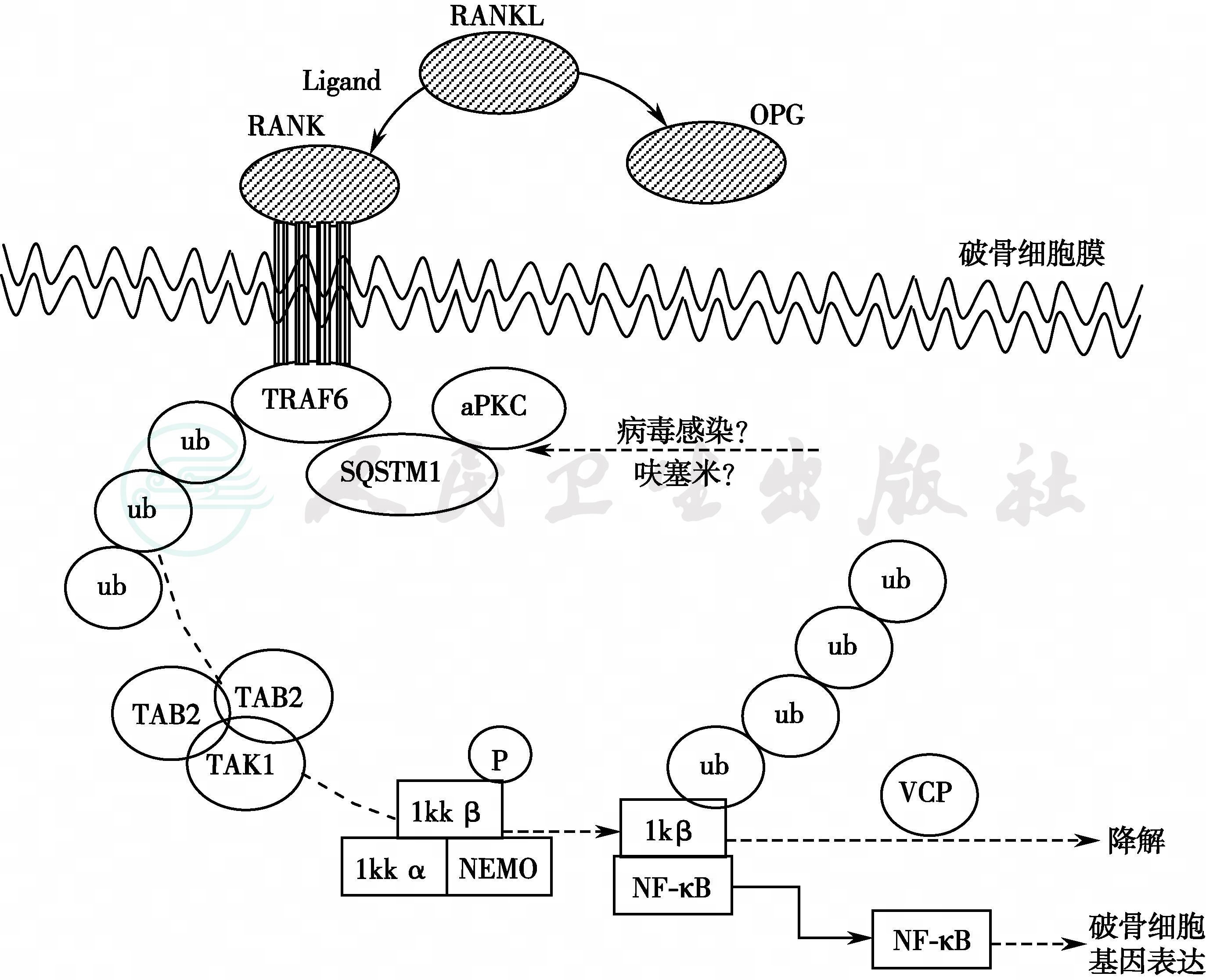
图1 RANKL-NF-κB信号途径
注:AANKL激活RANK,引起受体蛋白三聚化,受体的胞质尾部与适应蛋白TRAF6结合,并与非典型蛋白激酶C及SQSTM1形成复合物;TRAF6在泛肽连接酶的作用下,与多个小分子泛肽结合并形成多泛肽链。下游TAB2-TAB3适应蛋白识别并结合泛肽链,并进一步将IKK2-IKKβ-NEMO复合物激活(磷酸化),后者进入核内,选择性刺激破骨细胞基因表达。OPG可阻滞RANKL与RANK结合,而IκB的降解与VCP有关。病毒和其他环境因素如呋塞米诱发Page骨病的机制未明,可能与损害SQSTM1的正常功能有关。aPKC:atypical protein kinase,非典型性蛋白激酶;SQSTM1:sequestosome1
25%~30%的家族性和部分散发性PDB与sequestosome基因1(编码NF-κB途径中的支架蛋白)突变(如P392L)有关;有人认为,sequestosome 基因1突变可能只是成为PDB的一个易感因素而非直接病因。患者常伴有慢性副黏病毒感染(para myxoviral infection),表达麻疹病毒核壳基因(measles virus nucleocapsid gene)的转基因小鼠发生PDB样骨损害。
(二)VCP突变导致Paget骨病-肌病-痴呆三联征
SQSTM1基因突变是家族性PDB的常见病因,而散发性PDB的病因未明,涉及的遗传因素可能很多。近年发现,常染色体显性遗传性包涵体肌病(hereditary inclusion body myopathy)-Paget骨病(PDB)-额颞痴呆(frontotemporal dementia,FTD)症(IBMPFD,MIM 167320)是一种少见的高外显率的多系统性疾病,与散发性PDB有密切联系。IBMPFD以骨病-肌病-痴呆三联征为特点,其主要临床特点是:①肌病:属于包涵体肌病的一种,肌肉病变引起近端和远端肌无力(90%),常最先波及肩部和髋腰部肌肉,患者的乏力和虚弱呈进行性加重,不能抬腿升臂,上楼或登梯困难,肌肉酸痛,血清肌酶活性显著升高;最后可因呼吸肌与心肌病变而致死。②Paget骨病的临床表现与一般PDB相似,但发病年龄早,长骨和颅骨畸形及骨折常见,ALP和其他骨代谢标志物明显升高。③额颞痴呆的进展较快,由于大脑的额颞叶变性,语言和行为障碍特别明显,而撰写、绘画、计算和记忆力相对完好。少数患者伴有心肌病、肝纤维化、感觉和运动神经病变等。
IBMPFD是由于含空泡素蛋白(valosin-containing protein gene,VCP,CDC48,p97)突变所致,VCP是细胞代谢(cell metabolism)、胞膜融合(membrane fusion)、核膜重建(nuclear envelope reconstruction)、内质网相关物质降解(endoplasmic reticulum-associated degradation)、有丝分裂后高尔基池组装(post-mitotic Golgi cisternae reassembly)、泛素-蛋白酶体降解(ubiquitin-proteasome degradation)等过程的重要调节因子。目前,VCP突变引起IBMPFD的病例报道已有近200例,突变位点约16个。
骨病-肌病-痴呆三联征是诊断IBMPFD的必要条件,但一些患者可能不典型,其中以肌病的发生率最高(80%~90%),其次为PDB(43%~51%)和痴呆(30%~40%)。
(三)破骨细胞对RANKL和VD受体过敏感
Paget骨病的骨病理学特点是破骨细胞数目明显增多、活性增高。Kakita等发现,多核细胞具有Paget骨病破骨细胞的特征,表现为细胞成熟更快更多(高于正常10~100倍以上),细胞内核数目增多,抗酒石酸酸性磷酸酶表达显著升高等,电镜下可见呼吸道融合病毒核壳(nucleocupsid),但未发现胞核或胞质包涵体。Demulder等发现培养液中的CFUGM克隆生成明显增加。用高度纯化的生血前身细胞(CD34+细胞和CD34-细胞)共培养时,Paget骨病的破骨细胞前身细胞和CFU-GM克隆明显增多。由CFU-GM克隆衍生的破骨细胞所需要的1α,25-(OH)2D浓度仅为正常时的1/10,这说明Paget骨病患者的破骨细胞前身细胞对活性VD有过度反应。Menaa等进一步证明,Paget骨病患者的破骨细胞前身细胞由于RANKL的表达过多而使其分化增殖为成熟破骨细胞的潜能增高,即患者的破骨细胞对RANKL存在过度反应,而这种过度反应又与患者骨髓中的M-CSF、IL-6过高有关。
IL-6通过NF-κB信号途径导致破骨细胞的功能调节失常,因而可认为Paget骨病是一种与NF-κB信号调节失常有关的疾病。其过程大约是IL-6升高引起 RANKL分泌增加,从而导致破骨性谱系细胞(包括前身细胞和成熟破骨细胞)生成过多和破骨细胞活性增强。破骨细胞前身细胞对1,25-(OH)2D过敏感的原因不是这些细胞的维生素D受体(VDR)过多或VDR变异。用GST/VDR融合蛋白进一步发现,该类患者的破骨细胞前身细胞表达的TAFⅡ-20(TFⅡD家族成员中的一种)增多,其意义是:当TAFⅡ-20含量够多时,破骨细胞前身细胞的分化就不依赖于1,25-(OH)2D,所以在1,25-(OH)2D很低时,破骨细胞的生成仍是加速的。
本病的骨损害与骨肿瘤(尤其是多发性骨髓瘤)有许多共同特点,表现在:①病变局部的破骨细胞生成增多,骨吸收增强、加速;②导致破骨细胞数目增多和活性增强的原因相类似,都是过多IL-6和RANKL介导的结果;③病变的形态表现类似,而且Paget骨病易于并发骨肉瘤和其他骨肿瘤;④用二膦酸盐等药物治疗的效果较佳。只要能抑制破骨细胞的增生、分化和活性,就可达到控制骨病变之目的。
(一)炎症与异常新骨形成引起骨内压增大与骨痛
病变部位的破骨细胞在病变初期数量急剧增加,众多的异形破骨细胞聚集于Haversian管、骨内膜面及骨小梁的表面,破骨细胞数目增多,细胞变大,细胞核显著增多(正常时约2~3个核,本病时常多达数十个核或100个核以上),骨吸收活性明显增强。病程一般延续十天左右,此间通过抗破骨细胞药物治疗可控制病情。骨形成增加,但形成的新骨结构异常,主要为不规则的和不成熟的交织骨,呈骨痂状。随着病程的发展,新骨被重吸收,此时的破骨细胞在形态上也有明显异常。电镜下可见细胞内的微丝(microfilament)形如网状,位于核的附近或细胞质中。破骨细胞内的微丝生成和聚集为本病的特征性表现,并认为这是病毒感染的征象。此外,有时还可见到包涵体或病毒(样)核壳。在骨吸收部位有大量的成骨细胞聚集,骨形成亦相应增强。细胞内的粗面内质网、线粒体和Golgi发育良好。骨基质结构紊乱,可见大量的“嵌合样”外观的交织骨形成,黏合线不规则,基质与交织骨交替地混杂排列。骨小梁被破骨细胞侵袭,邻近吸收区的髓腔内有板层骨和交织骨形成的新骨带,偶尔可见血性囊肿,并出现含铁血黄素的巨噬细胞。该病累及的程度和部位具有明显的不均一性。常见多个部位受累,最常见的部位有骨盆、腰椎和股骨。95%以上的患者初期无明显症状,仅X线证实为Paget骨病。除骨折外,该病的发作具有隐袭性,30%的患者有长达10年的骨病症状,难以与其他骨关节疾病的骨病相区分。
多数患者临床表现不明显,当有并发症而进行X线检查或检测血ALP时才被意外发现。本病的常见主诉是骨痛,表现为局部病灶的固定性钝痛,呈烧灼感,常在夜晚发作或加重。偶尔出现锐痛或放射性疼痛。骨痛的机制不明,可能与骨膜膨胀或骨髓充血刺激感觉神经有关。疼痛也可能是并发症所致,如关节退行性变和钙化性血管周围炎等。负重可使下肢、脊椎和骨盆的疼痛加重。严重骨痛者在局部往往可发现体温上升、骨内压增大,伴疲劳无力甚至衰竭或嗜睡。早期的颅骨病变为额骨和枕骨的局部溶骨性病变,骨髓透过缺损的骨外板而使病变部呈紫红色,但无疼痛或其他症状。高钙血症和高钙尿症仅见于一些病变广泛和长期活动者,部分可能与合并原发性甲旁亢有关。有些患者因尿酸过多可导致高尿酸血症,出现肾石病。
Paget骨病累及骨骼达30%以上时,或单独累及颅骨时,可出现心排出量增加。重症者常并发心瓣膜钙化及相关病变。主动脉狭窄达30%,完全性房室传导阻滞、不完全性房室传导阻滞、束支传导阻滞和左室肥厚的发生率分别为11%、11%、20%和13%。重度颅底陷入时可伴有动脉“盗血”综合征。Paget 骨病患者的动脉硬化性钙化的程度与范围均明显比同龄正常人严重,Paget 骨病与动脉硬化存在一定的病理生理联系。
(二)骨增生硬化与狭窄导致骨畸形伴听觉及骨关节功能障碍
破骨细胞开始减少,作用衰减;成骨细胞增多聚集在骨表面,形成新的板骨层,呈镶嵌图形。不整齐的锯状板层骨互相叠加,表明骨吸收和骨形成交替进行,无法保存完整的骨单位,邻近的骨髓逐渐为结缔组织所代替。
颅骨吸收阶段可持续多年,颅骨周径逐渐增大,可伴有感觉神经性听觉障碍。颅骨外板增生可引起颅底孔道变窄,压迫脑神经,其好发部位为颞骨岩部,故常合并听神经功能障碍,导致感觉性听力丧失、中耳骨化和慢性炎症等病变,或导致视乳头水肿、眼肌病变、突眼,视神经萎缩及失明。后期导致头痛、痴呆、脑干或小脑功能障碍。累及脊椎(主要发生在腰骶段)时,引起腰痛,与局部的骨损害、脊髓压迫性损伤、骨质疏松及脊椎关节炎症、硬脊膜外脂肪钙化、脊椎局部软组织增生、局部出血、脊椎骨折后的肉瘤样变性(sarcomatous degeneration)等有关;持续性腰痛的另一个原因是脊髓缺血,与病变形成过多的动脉侧支循环(称为动脉盗血综合征,arterial steal syndrome)有关。腰椎侧弯,脊髓受压,少数患者甚至逐渐出现下肢麻木乃至痉挛性瘫痪;波及颈椎可出现颈椎脱位,累及股骨和胫骨等下肢长骨可出现膝内翻、下肢外旋、胫骨向前向外弯曲、髋或膝关节活动明显受限;肢体长短不对称,关节变硬,张力增加,外伤后愈合困难,易形成骨折。骨盆骨病变早期的症状不明显,只是达硬化阶段才有耻骨加宽、髋臼内陷和髋关节活动受限。
骨皮质加厚,骨髓腔阻塞,新生骨硬而脆,股骨和胫骨常呈侧弯或前弯畸形;颅骨外板出现增生性硬化病灶,外形明显增大、畸形。骨盆受累可导致髋臼突出(髋关节内陷)。Paget骨病的骨代谢十分活跃,骨重建量可达正常的10倍以上(正常的骨重建单位约为10%或更少),病变骨和正常骨的结构分界明显。骨体积增加,常伴有微骨折和畸形,新生骨脆性增加,胶原纤维排列紊乱,板层结构不稳定,容易折断。从病程发展上看,同一病灶中上述3个阶段并非绝对分开,很多情况下同步或交错发生,少数病灶可有癌变。伴甲状旁腺功能亢进时,有不规则板层骨碎片,绕以纤维性骨炎,或有棕色瘤及囊性骨炎,但骨黏合线很少。骨折后的骨痂有板层骨结构及黏合线,但骨单位完整,新骨形成规则。
(三)部分患者的病情反复并可恶性变
骨关节病变主要有关节畸形、退行性关节病变、软骨钙盐沉积、假性痛风、钙化性关节周围炎等。骨折有三种类型,即裂纹骨折、长骨断裂和椎体压缩性骨折。可在轻微外伤或无外伤情况下发生,骨折不愈合率达40%。骨病变畸形可能导致关节畸形,但Paget骨病本身很少侵犯关节软骨面;当骨畸形累及髋关节相邻部位时,因运动应力异常可导致关节异常磨损,软骨缺损,而下层出现假血管瘤样物,晚期出现髋臼内陷。膝关节也有类似情况,在远离病灶的部位可出现钙化。
多发性骨肉瘤样变性病变是Paget骨病的最严重并发症之一,病变可发生于任何部位,多见于老年病例。多数为骨肉瘤,亦可为纤维肉瘤或其他类型的肉瘤,继发性骨巨细胞瘤或合并骨巨细胞瘤者(多为良性,偶为恶性)少见。Paget骨病合并骨肉瘤(Paget骨肉瘤)主要发生于Paget骨病的老年患者伴多骨损害时。Paget骨肉瘤应与转移性骨肿瘤鉴别。Paget骨病患者发生骨肉瘤的概率为正常人的数千倍以上,其发病机制未明。有人认为与染色体18q20-21体质性杂合子丢失(loss of constitutional heterozygosity,LOH)有关。在染色体18q的18S60和18S42之间含有肿瘤抑制位点,丢失这些位点可导致骨肉瘤的发生。同时,这一部位也是家族性Paget骨病和家族性扩张性骨溶解(familial expansile osteolysis,FEO)的基因位点,故Paget骨病易并发骨肉瘤。伴发肉瘤的患者有骨痛、肿胀和病理性骨折,其预后差,化疗和手术仅能控制症状,而对病变本身无明显疗效。放射治疗和截肢可减轻疼痛。术后5年的存活率为5%~8%。
(一)碱性磷酸酶急剧增高提示病情恶化或骨肉瘤
15%~20%的患者因骨重建对钙的需求增加,血钙廓清加速导致血PTH上升。骨受累部位广泛的患者或合并原发性甲旁亢时有高血钙症和高尿钙症。血ALP水平与病变范围和病变的活动程度有关。体积小的骨骼病变(约10%)ALP正常。颅骨病变时ALP升高。如并发骨肉瘤,ALP可急剧增高,酸性磷酸酶和5-核苷酸酶也可升高。正常人在低明胶饮食时的尿羟脯氨酸的排泄量低于50mg/d,而Paget骨病患者因其骨重建旺盛,尿羟脯氨酸排泄量可高达2000mg/d。此外,尿羟赖氨酸也能反映骨重建活动的水平和本病的病变程度。血cathepsin K也是反映破骨细胞功能的良好指标。
(二)骨吸收和骨硬化常互相转化
早期为溶骨期,X线片上可见界限分明的圆形局限性骨质疏松区;第二阶段为不匀称的骨溶解和硬化表现;骨质硬化为第三阶段的突出表现,皮质骨增厚,骨小梁增粗,骨病变部位常呈海绵状改变或为紊乱结构象,两种情况可单独或同时出现。海绵状结构较为常见,骨质粗糙、骨干增宽、骨小梁紊乱。骨皮质被海绵状结构所代替,骨髓腔和骨皮质间的界限不清;广泛不规则的骨质致密,或匀称一致如粉笔状、颗粒状或灰浆样。X线表现可反映疾病的病理变化过程。X线平片征象大致有骨质吸收、骨质硬化及两者的混合型3种类型。同一病例或同一病变内不同类型的病变可互相转化。早期X线表现为骨质溶解吸收,以颅骨明显。
1.颅骨
受累时表现为局限性BMD降低,起始于外板,疏松部的边缘光滑;有时可发展到整个颅骨穹隆。后期出现片状骨硬化,主要发生于内板,出现皮质增厚,内、外板失去正常分界。颅基部受累时,颅底内陷,蝶鞍变小,不规则。上颌受累较下颌骨多见,导致面部畸形。开始为颅骨外板向内板蔓延的“局限性骨质疏松”,其边界清晰。此后由于骨质吸收区过度修复,形成骨质硬化或粗糙的骨小梁。在颅骨则表现为“棉花球”状致密影。此后局限性骨质吸收与骨质硬化病变混合存在,颅骨内、外板界限消失,颅骨增厚,有“骨性狮面”及扁平颅底或颅底凹陷征(图2)。
2.骨盆
约2/3的患者可有骨盆病变,多表现为BMD减低和畸形,可伴不规则囊性变低密度囊状透明影与灰浆状高密度病变混杂存在、或髋臼稍内陷;核素骨显像可见“鼠面征”及放射性摄取增高(图3)。骨盆入口为三角形,股骨头变形致髋内翻畸形或髋臼突出,髋关节间隙狭小(应与退行性关节炎相区别),外上象限负重区狭窄。骨盆变形呈“香炉状”,表现为髂骨翼外翻、骨盆口呈三角形、髋臼及股骨头内陷。骨盆的髂耻线增厚表现为“碗边征”。
3.长骨
以股骨、胫骨和肱骨受累多见。早期的典型表现为“V”形吸收区,皮质骨呈非对称性膨胀,病灶溶骨,长度增长,呈弓状畸形。骨小梁纹理粗乱,骨髓腔硬化狭窄。骨膜下有完全或不完全裂纹性骨折。长骨病变的早期可呈“草叶样”改变,在病变与正常骨质间有“V-型骨质稀疏区”,长骨弯曲变形,凸面易发生不完全骨折,骨折线与骨干长轴垂直,愈合延迟;凹面骨皮质则多增厚、致密,有时可使骨髓腔变窄或闭塞。
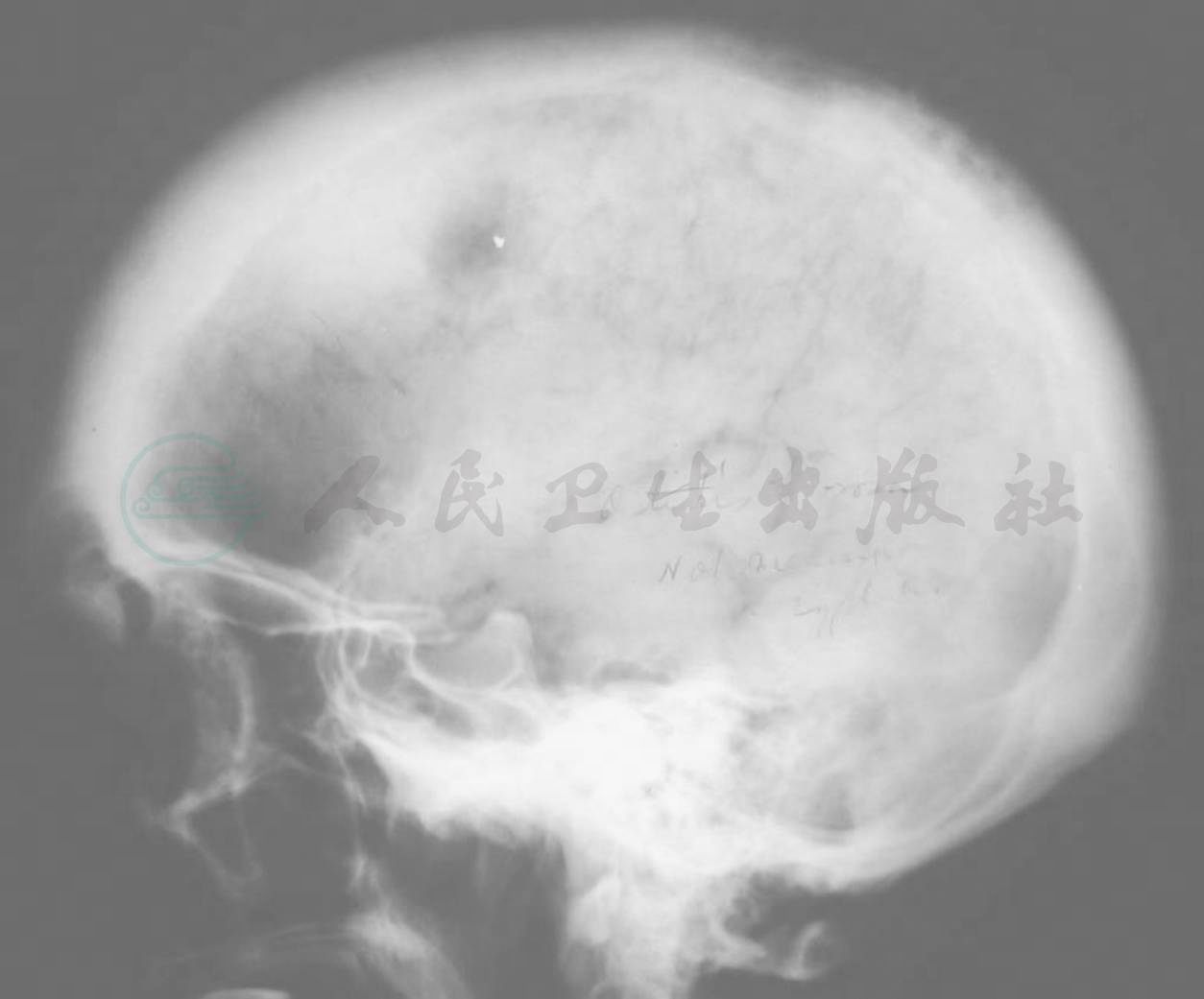
图2 Paget骨病(颅骨侧位片)
注:男,73岁,颅骨侧位平片。颅板增厚,板障消失,颅缝不能显示,颅穹窿部不规则形态的灰浆状高密度病变与低密度透明区混杂存在,颅底凹陷
4.脊椎
受累时表现为椎体中央粗糙,如栅栏状,边缘变厚;也可表现为椎体中心和两侧横突密度增高,由3个点状影构成一倒置三角形者被称为“鼠面(mouse face)”征(图4),可分为Ⅰ度和Ⅱ度。“鼠面征”对Paget骨病的诊断有重要意义。随后椎体增大,并出现压缩性骨折。椎体被压缩变形,椎体周围骨皮质增厚,表现为“框样椎体”。
短期治疗的目的为降低骨转换和缓解症状,降低神经系统并发症、骨骼畸形、改善听力;长期治疗的目的是防止骨关节炎、诱导缓解、防止疾病进一步发展,降低致残率。
(一)一般治疗
Paget病的疼痛治疗可采用非甾体消炎药(NSAIDS)或COX-2抑制剂。小剂量三环类抗抑郁药对部分患者有效。由于骨关节炎或神经根压迫引起的疼痛可采用阿片类镇痛剂、针灸、电刺激神经疗法、水疗、关节置换、手术或辅助行走器治疗。
由于Paget病患者骨形成增加,因此患者需补钙1000~1500mg/d、VD 400~800IU/d,特别是对于经过二膦酸盐或降钙素治疗的患者常并发继发性甲旁亢,个别甚至发生三发性甲旁亢,补充钙剂和VD采用尤为重要。
(二)抗骨吸收药物治疗
大部分患者症状轻微或无症状,无需治疗。患者多因疼痛、畸形、活动困难或因并发症(如骨折、肉瘤样变及其他器官继发性病变等)而就诊。药物治疗的前提是:①患者疼痛剧烈,经X线和核素检查能明确病变者;②心功能衰竭或心排出量明显增高;③高钙血症和因高尿钙而导致反复发作肾石病者;④骨的代谢转换率升高和骨病变明显者。曾用于本病治疗的药物有降钙素、普卡霉素(plicamycin)、胰高糖素、放线菌素D、依普黄酮(ipriflavone)和硝酸镓(gallium nitrate)等,这些药物主要是抑制破骨细胞活性,但这些药物均为二膦酸盐所取代,二膦酸盐为抗骨吸收药物,对早期的骨痛、骨内压增大、骨吸收亢进和中期的镶嵌状板层骨增生并骨畸形有特殊疗效,但应慎用或禁用于晚期的骨硬化,特别是伴有明显的颅底和颈椎骨增生硬化者,因为有可能加重神经压迫或导致颈椎骨折或移位等严重并发症。
1.二膦酸盐
当病变骨出现纤维发育不良(fibrous dysplasia)或溶骨性损害时,均可使用二膦酸盐制剂,可抑制骨和软组织钙化,阻滞骨吸收和破骨细胞增殖。
FDA批准用于Paget病治疗的二膦酸盐见表2。根据2000年西方骨质疏松联盟会议的建议,etidronate不宜再用于本病的治疗,要尽量使用第二或第三代二膦酸盐(如alendronate、pamidronate),因为它们改善骨病变、纠正骨代谢异常的作用明显强于etidronate。pamidronate的优点是作用强,药效时限长,短程治疗可缓解达数月之久,但抑制骨矿化作用也大。其不良反应是首次服用或注射后会有低热,有轻度暂时性低钙血症、低磷血症和淋巴细胞减少等。同时口服元素钙500mg/d可预防和减轻低钙血症和继发性高PTH血症。一般用量是30~90mg加入0.9%生理盐水或5%葡萄糖250~500ml中静滴1~4小时以上。在静注溶液中本药浓度不应大于15mg/125ml,每个疗程最大总量为90mg,治疗总量取决于治疗前的血钙浓度。与传统的鲑鱼降钙素和依替膦酸二钠的作用相比,alendronate对骨重建生化指标的下降作用仅为80%,但缓解期可达1年以上。其不良反应是恶心、消化道不适等。建议剂量为40mg/d,早晨空腹,避免卧位,200~300ml白开水送服。服药后30分钟进食,同服元素钙500mg/d以防止低钙血症。6个月为1疗程,1年后指标升高时再复治。
表2 FDA批准用于Paget病治疗的二膦酸盐

替鲁膦酸钠(tiludronate)和利塞膦酸盐(risedronate)的建议剂量为30mg/d,连服84天后停药。与依替膦酸二钠比较,本药的主要优点是疗程短,缓解期长,止痛效果明显。
在药物选用中应注意,依替膦酸二钠效果稳定,患者依从性好。帕米二膦酸盐对病情较轻的患者效果佳,静脉滴注1次可维持疗效1年,重症患者需多次滴注。新近的1项研究表明,当氨基二膦酸盐在临床使用中产生抗体时,用同类的药物(如阿仑膦酸盐,alendronate)仍能缓解病症。当上述二膦酸盐制剂的疗效不佳时,建议改用作用最强的唑来膦酸盐(zoledronate),临床研究经证实,唑来膦酸盐可有效降低患者ALP并缓解症状,但目前应用于儿童患者的经验缺乏。
奈立膦酸(neridronic acid)为含氮二膦酸盐的一种衍生物,其作用机制与其他二膦酸盐相似,但奈立膦酸与正在进行骨重建骨组织的亲和力最高。用于治疗PDB时,可静脉注射200mg后,完全缓解率65%,骨转换率下降75%,一般仅单用一次。但是,二膦酸盐可以抑制骨吸收,但不能预防病变复发。
2.降钙素
一般不作为本病治疗的常规首选药物,因其不能使破骨细胞凋亡。一般仅在缺乏二膦酸盐或需要紧急控制症状(如手术中)时使用。降钙素的主要作用是延缓骨吸收,迅速抑制破骨细胞的功能并减少其数量。一般可用鲑鱼降钙素(salmon calcitonin;商品名:密钙息)每次100U,每周3~4次;4周后改为每次50U维持,每周3次。鼻腔喷药和直肠栓剂有相同效果,注意测定血ALP和尿羟脯氨酸水平。4~8周后判断其治疗效果。治疗标准为骨的代谢转换率降至正常而不出现明显的低钙血症。儿童使用降钙素后,因骨吸收受抑制较成人快,同时尿钙、磷、钠和尿酸排泄增加,胃酸和胰液分泌下降,小肠电解质分泌增加,易出现低钙血症。
(三)严重难治性Paget骨病试用普卡霉素治疗
普卡霉素(光辉霉素,mithramycin)为细胞毒性抗生素,仅用于严重难治性Paget骨病。该药能抑制RNA和蛋白质合成,同时也抑制前破骨细胞的活性,缓解疼痛。有效剂量为每天15~25μg/kg,一般静脉使用10天。主要不良反应有肝毒性和骨髓抑制等,常和地塞米松合用。
(四)手术治疗骨折/脊髓-神经根受压/严重畸形/骨肉瘤
手术治疗的适用证是:①多发性骨折;②需要长期固定者;③脊椎或神经根受压;④严重畸形;⑤预防或处置肉瘤。手术治疗时,必须配合应用相应的二膦酸盐药物治疗。
在上述条件下,颅底陷入、瘫痪、四肢畸形和病理性骨折的手术治疗通常需要使用降钙素等来确保手术的安全性和减少术后并发症。为减少术中出血,促进术后伤口愈合,一般在手术前应服用1周至数个月的药物。合并复发性肾结石、高钙血症、痛风、充血性心力衰竭的患者更应在术前接受一段时间的药物治疗。严重畸形应于病情稳定时及时进行截骨术治疗。骨折发生时,应一并纠正骨畸形。合并脊髓压迫症状时可进行椎体截除减压术。对恶性变者应及时行截肢术,按继发性恶性骨肿瘤进行综合治疗。









