


 收藏
收藏 已收藏
已收藏英文名称 :heredi tary vitamin D‐resistant rickets
遗传性VD抵抗性佝偻病(或骨软化症)(heredi tary vitamin D‐resistant rickets,HVDRR)有两种类型:Ⅰ型缺陷是由于肾脏1α‐羟化酶缺陷,使1,25‐(OH)2D3合成减少。此型使用大剂量天然VD治疗有效,用生理剂量的1,25‐(OH)2D3或1α‐(OH)D3治疗亦有一定疗效。Ⅱ型的缺陷在VDR对1,25‐(OH)2D3作用有抵抗,又称遗传性1,25‐(OH)2D3抵抗性佝偻病(HVDRR)、VD抵抗性佝偻病Ⅱ型(vitamin D resistant rickets typeⅡ)、假性VD缺乏性佝偻病Ⅱ型(pseudovitamin D deficiency ricketsⅡ,PDDR‐Ⅱ)、钙三醇抵抗性佝偻病(calcitriol‐resistant rickets)、VD抵抗性佝偻病(vitamin D‐resistant rickets)、遗传性低血钙性VD抵抗性佝偻病(hereditary hypocalcemic vitamin D‐resistant rickets)等。因该综合征是由于对VD遗传性抵抗所致,故文献多认为该综合征应命名为遗传性VD抵抗性佝偻病(hereditary vitamin D‐resistant rickets,HVDRR)。
HVDRR于1978年由Brooks等首先报告。其特点为:①有佝偻病或骨软化的临床表现,常于出生后不久发病;病人有骨痛、肌无力、肌张力下降或低血钙性手足搐搦;患儿生长发育延迟,牙齿发育停滞等。可因并发肺炎等疾病而死亡。有些患儿头发稀少或全秃(包括眉毛缺如)。②血清中1,25‐(OH)2D3升高。③低钙、低磷血症,伴血清碱性磷酸和PTH升高,血清25‐(OH)D3正常,但1,25‐(OH)2D3明显升高,24,25‐(OH)2D3正常或下降,这些指标在使用大剂量VD后不能得到改善。④继发性甲旁亢。⑤全秃。⑥有家族发病倾向,呈常染色体隐性遗传。本病罕见,迄今世界文献中已报告的病例约数十例。
本病为遗传性疾病,但遗传缺陷存在不均一性。遗传方式为常染色体隐性遗传,发病呈家族性,男女发病几率相等,且常一个家庭有几个患儿。患者父母近亲结婚者多,无临床症状且骨骼发育正常,多为表型正常的杂合子,患者则为纯合子。其病因为VDR基因突变,导致VDR功能异常。
(一)激素与受体结合缺陷
这一类缺陷包括VDR数目减少和VDR对1,25‐(OH)2D3的亲和力降低,VDR受体蛋白结合1,25‐(OH)2D3的量减少或缺如。例如Ritchie等报告无亲缘关系的4例病人,在C末端外显子7的第970位胞嘧啶突变为腺嘌呤(TAC→TAA),编码的VDR氨基酸密码子(TAC)变为终止密码子,VDR基因提前终止转录,使VDR蛋白被截去292个氨基酸,其中包括VDR的配体结合区(ligand‐binding domain,LBD)在内,使VDR分子量减小(正常为50ku),也有被截去291和362个氨基酸者,结果VDR不与1,25‐(OH)2D3结合,称之为VDR结合阴性型。VDR的LBD区的常见突变类型见图1和图2。患者为纯合子,其父母为杂合子,如R30stop突变型VDR缺失398个氨基酸残基(包括锌指结构的大部分及全部激素结合结构域),这种病例的病情往往更为严重。Malloy报道一例亚洲病人,临床表现典型,于第6外显子发现错义突变(T→G),编码的蛋白251位氨基酸苯丙氨酸变为半胱氨酸,使得VDR数量及亲和力均下降,干扰RXRα异二聚体形成而导致发病。另一例HVDRR患者亦具备该病各临床特点,且严重体秃,基因测序示第8外显子点突变(C→T),使得蛋白产物317位氨基酸密码子变为终止密码子,导致VDR的LBD缺失110个氨基酸,VDR不能与1,25‐(OH)2D3结合而发病。
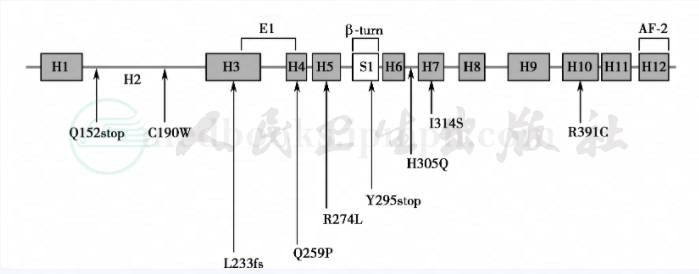
图1 VDR蛋白的配体结合(LBD)区和该区的部分突变位点
LBD区突变导致配体结合阴性反应。图中的H1~H12代表VDR蛋白配体结合区的α‐螺旋;S1代表β‐折叠区;fs代表读码框架(framesheft)

图2 VDR的配体结合区及其突变位点
图中箭头所示为点突变位点,H1~H12代表α‐螺旋区,β‐turn为β折叠处。图中所标出的突变均导致HVDRR
(二)VDR复合物与DNA结合缺陷
VDR结合1,25‐(OH)2D3的量与亲和力正常,但与特异性DNA反应元件结合有缺陷,使1,25‐(OH)2D3定位到细胞核中的量减少。DNA的反应元件含有两个锌指,锌指中如有突变,则引起锌指功能异常。故此类突变多见于外显子2和3(图3)。文献中已报告的锌指区点突变类型有:①VDR基因第一锌指顶端突变(如GGC→GAC突变导致在VDR中的甘氨酸被门冬氨酸取代)。②两锌指间的内含子突变(如CGG→CAG突变使VDR中的精氨酸被谷氨酸取代)。③第二锌指顶端突变(如CGA→CAA突变导致VDR中的一个精氨酸被谷氨酸取代)。④第二锌指的底部突变(如CAC→CGG或点突变导致与上述相似的氨基酸突变)。⑤VDR基因在外显子3突变(如第146位鸟嘌呤突变为腺嘌呤,使VDR的第47位的精氨酸被谷氨酰取代)。前述VDR基因的突变也使VDR电荷发生变化,从而导致VDR对DNA的亲和力下降,提示VDR复合物不能进入细胞核中与DNA结合。这种缺陷也是由于VDR基因突变所致降低,进一步使1,25‐(OH)2D3与DNA结合减少(见图3)。Malloy报道一例HVDRR患者,临床表现典型,但无秃头,行序列分析后发现VDR基因第7外显子上点突变(T→C),相应蛋白产物的268位氨基酸异亮氨酸由苏氨酸占据,导致与1,25‐(OH)2D3亲和力较野生基因型下降80%~90%。其报道的另一HVDRR患者,亦无秃头表现,于第4外显子发现5bp缺失/8bp插入,则LBD缺失2个氨基酸(H141 and T142)/插入3个氨基酸(L141、W142和A143),突变VDR活性较野生基因型减低1000倍而发病。

图3 VDR的DNA结合区(DBD)及其部分突变位点的分布
(三)核定位缺陷
病人的成纤维细胞可溶性提取物与1,25‐(OH)2D3有正常的或近乎正常的结合,但在完整细胞中未能检出1,25‐(OH)2D3定位于细胞核,如:①ATC→AGC(I 314 S)突变;②CGC→TGC(R 391 C)突变。推测这些病人存在有对核定位有重要作用的二聚体化有缺陷。
(四)VDR后缺陷
VDR结构和功能以及与DNA结合反应均正常,但诱导24,25‐(OH)2D3生成减少。正常人皮肤成纤维细胞与1,25‐(OH)2D3同时温育8小时,可使加入媒液中的25‐(OH)D3转化为24,25‐(OH)2D3的量比未加1,25‐(OH)2D3的对照细胞的量增加20倍,有此缺陷的病人则明显减少(0.11~0.27pmol·min-1·mg-1蛋白对0.02pmol·min-1·mg-1蛋白)。
BAG‐1 P50、‐P46、‐P33、‐P29为人体中的四种抗细胞凋亡蛋白的异构体。BAG‐1 P46与类固醇类激素受体结合并调节其活性,BAG‐1 P50可与VDR相互作用,或促进VDR与DNA反应元件结合,继而抑制靶基因的转录,细胞生长加速,且阻滞了1,25‐(OH)2D3诱导的细胞增殖作用,使1,25‐(OH)2D3不能诱导VDR的活性,因此BAG‐1 P50是一种VD信号转导途径的调节因子,如果表达过度可导致靶细胞对1,25‐(OH)2D3治疗的抵抗。一个经典的点突变即为VDR的LBD第420位谷氨酸由赖氨酸替代(E420K),与甾体激素受体共激活剂‐1(SRC‐1)及DRIP205结合功能受损从而导致本病的发生。
1.Ⅰ型:①肾脏1α‐羟化酶缺陷;②1,25‐(OH)2D3合成减少;③大剂量维生素D治疗有效。
2.Ⅱ型:①维生素D受体突变;②遗传性1,25‐(OH)2D3抵抗性佝偻病(HVDRR);③大剂量维生素D治疗无效或疗效很差。
1.血液生化:①血钙低、ALP升高、骨源性ALP增高;②血清磷正常或降低、PTH升高。
2.尿液检查:①尿钙减少、cAMP增高;②氨基酸尿;③尿磷正常或增高;④尿羟脯氨酸增高。
3.血清维生素D: ①25(OH)D3正常;②1,25‐(OH)2D3明显升高;③24,25‐(OH)2D3正常或稍低。
4.骨骼X线检查:①普通性脱钙;②佝偻病表现。
5.维生素D受体基因突变。
1.维生素D: ①选用维生素D2或D3、25‐(OH)D3、1,25‐(OH)2D3及其类似物;②维生素D ∶24,25‐(OH)2D3∶1,25‐(OH)2D3的效力比值为1 ∶10 ∶1000。
2.1α,25(OH)2D3 :①剂量应因人而异,以纠正低血钙为度;②补充钙剂。









