


 收藏
收藏 已收藏
已收藏FGF-23是一种利尿磷因子,在高血磷和1,25-(OH)2D升高情况下,骨细胞和成骨细胞分泌的FGF-23增多。FGF-23基因突变、GALNT3基因突变(影响FGF-23翻译后修饰)或klotho(FGF受体1转换为FGF-23受体的辅助因子)突变引起严重的低磷血症和肿瘤样钙盐沉着症。
1.血FGF-23升高
在FGF-23分泌过程中,分子C末端的179~180位氨基酸被裂解,如果FGF-23的RXXR弗林蛋白酶(成对碱性氨基酸蛋白酶)样裂解结构域(RXXR furinlike cleavage domain)突变(R176Q、R179W 等),FGF-23 不能被灭活,引起活性FGF-23显著升高,导致低磷血症性佝偻病。虽然最初的研究发现,PHEX组装FGF-23,但以后的研究并未证实FGF-23的裂解依赖于PHEX,因此弗林蛋白酶(furin)结构域突变是ADHR的合理解释,详见本篇扩展资源12相关内容。据报道,完整的血清FGF-23(intact FGF-23)浓度为(44±37)pg/ml,但受年龄、性别、体重和肾小球滤过率的影响。慢性肾病患者的血清FGF-23明显升高,并且是心血管事件的预报因子。肿瘤引起的低磷血症和X-性连锁遗传性低磷血症患者血清FGF-23亦明显升高,切除肿瘤后下降。由于其他原因所致的低磷血症患者的血清FGF-23显著降低,多数监测不到(低于3pg/ml)。血清FGF-23明显升高伴低磷血症提示其病因为FGF-23分泌过多。FGF-23升高引起佝偻病/骨质软化症的共同特点是肾脏磷的消耗和1,25-(OH)2D的不适当降低,原因是骨细胞生成的FGF-23过多、肿瘤或骨纤维样发育不良分泌过多FGF-23或FGF-23降解缺陷。
(1)慢性肾病:
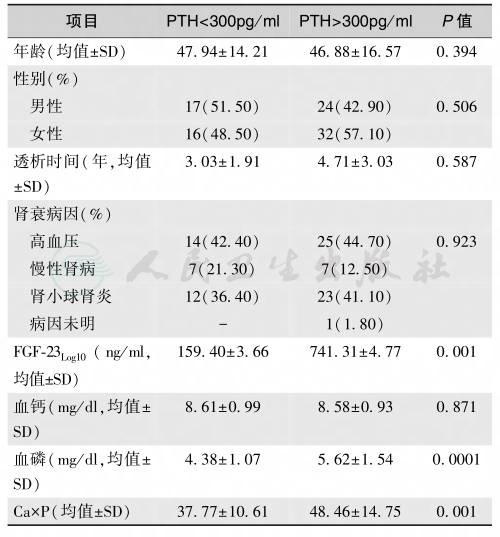
在慢性肾病患者中,血清FGF-23升高与顽固性继发性甲旁亢(血清PTH>300pg/ml)及血清钙磷乘积相关,而与其他临床指标无关,提示FGF-23水平是评价肾病严重程度的重要指标(表1)。
表1慢性肾病血透患者血清FGF-23与PTH、血钙磷乘积的关系
(2)X-性连锁低磷血症性佝偻病(XLH):
PHEX突变引起FGF-23降解缺陷,血清FGF-23升高。
(3)常染色体隐性遗传性低磷血症性佝偻病:
细胞外基质蛋白家族中的小分子整合素-结合配基N-连接的糖蛋白DMP1突变,使骨细胞中的FGF-23转录增加,骨矿化缺陷。
(4)肿瘤和骨纤维发育不良:
McCune-Albright综合征的病因为GNAS1的活化性突变,部分患者因骨细胞合成与分泌的FGF-23增多,引起高FGF-23血症。某些肿瘤因表达MEPE和sFRP4过多而导致PHEX和DMP1增加,使血FGF-23升高。
(5)原发性甲旁亢:
患者可表现为甲状旁腺腺瘤或增生,其病因与α-klotho近端的裂解点(breakpoint)易位,使β-葡萄糖苷酶(β-glucuronidase)编码障碍,这些病例的特点是血磷降低,血klotho和FGF-23显著升高,而PTH可能正常或仅轻度升高。
(6)FGF受体突变:
FGF受体亚型1/3/4突变使FGF-23不能与受体结合,通过受体调节使血FGF-23升高,并可引起低磷血症。
(7)线型脂肪痣或表皮痣综合征:
线型脂肪痣或表皮痣综合征(linear sebaceous/epidermal nevus syndrome,ENS)的表皮细胞的FGF受体3活化性突变引起表皮痣综合征,皮损呈线状、骨量减少伴低磷血症性佝偻病,同侧局限性骨病变伴表皮痣和血FGF-23升高为本病的特征。表皮痣综合征属于骨颅发育不良(osteoglophonic dysplasia,OD)中的一种,病因为FGF受体1、2或3突变,患者伴有颅缝早闭、眶上嵴前突、鼻梁下陷和肢根短小畸形。
2.血FGF-23降低
引起FGF-23降解减少的主要原因有常染色体显性低磷血症性佝偻病(ADHR)。









