


 收藏
收藏 已收藏
已收藏维生素D缺乏与不足尚无明确定义,原因是国际上对其评价方法和界定值(尤其是正常维生素D营养状态的切点)仍有争论[1,2]。如果以评估维生素D营养供应的公认指标——血清25-(OH)D(半衰期约3周)为标准,可将维生素D的营养状态分为典型维生素D缺乏、轻度维生素D缺乏、维生素D不足和维生素D正常四种。全美第三次健康和营养调查资料显示,多数居民的血清25-(OH)D低于正常,并随着年龄增长而下降,黑种人降低更明显;65岁时髋部BMD降至峰值骨量的50%,85岁时相当于10岁儿童水平。看来,维生素D的饮食推荐量(DRI)应该重新制订。20世纪是科技、信息、交通和移民空前发展的一个时代,环境污染日益严重,阳光照射越来越少,维生素D缺乏/不足在全球流行;甚至在阳光充足的巴西 Sao Paulo,血清25-(OH)D<50nmol/L 或<20ng/ml的人群亦分布高达71.2%及55.8%;继发性甲旁亢的发病率达到61.7%和54.0%。近年有关佝偻病/骨质软化症研究的主要重点在于维生素D的流行病学调查和单基因突变所致的慢性低磷血症性综合征。
调查显示,在一般人群中,维生素D不足的发生率为30%~50%,全球有近10亿人的维生素D缺乏或不足。据报道,42%的15~49岁美国女性黑人、40%以上的美国和欧洲老人、36%的成人患有维生素D缺乏/不足,另有50%正在进行骨质疏松治疗的绝经后妇女维生素D低于30ng/ml。在亚洲,东亚和东南亚地区,绝经后妇女维生素D不足的患病率>45%。随年龄增加,血浆25-(OH)D水平下降。导致与年龄有关的血浆25-(OH)D水平降低的原因有:①进食含维生素D食物量不足;②小肠维生素D吸收障碍;③接受日光照射较少,老龄亦影响7-脱氢胆固醇转化为维生素D的光化作用;④25-(OH)D代谢清除率增加。人类由脊椎动物进化而来,在漫长的进化过程中,人类的生活生产习惯有力巨大改变,如衣着、住房、室内生活和工作等,但相对于基因组,特别是调节维生素D代谢和功能的基因群对环境的适应性来说,这种进化仍然是迅速的,因此,人体内的抗维生素D缺乏机制是不完善的,特别容易发生维生素D缺乏或不足。我国大部分地区人群从食物中摄取的维生素D均难以满足需要,日光照射量也直接影响体内维生素D水平。阳光照射受户外活动时间、房屋光照、衣着、季节气候和大气污染等因素影响。长期在阴暗、烟雾过多、阳光照射不足的环境中工作生活者,常需额外补充维生素D制剂。
典型维生素D缺乏者出现佝偻病或骨质软化症,血清25-(OH)D 水平≤10ng/ml(25nmol/L,由 ng/ml换算成nmol/L时,乘以系数 2.5);血清25-(OH)D在11~20ng/ml(27.5~50nmol/L)范围时,表现为轻度维生素D缺乏/不足,可以导致骨质疏松症,但一般以骨骼外病变为主;维生素D不足时的血清25-(OH)D水平为20~30ng/ml(50~75nmol/L)。维生素D缺乏和维生素D不足统称低维生素D状态(hypovitaminosis D)。维生素D充足是指能维持骨骼和骨骼外组织健康的血清25-(OH)D水平(一般定为≥30ng/ml,有的地区或单位定为≥20ng/ml)。血清25-(OH)D≥150ng/ml(375nmol/L)称为高维生素 D 状态(hypervitaminosis D),见于维生素D过量和维生素D中毒两种临床情况。建议将正常血清25-(OH)D)的切点定为30ng/ml的理由是,该水平维生素D能防止发生继发性甲旁亢以及确保肠钙吸收和骨骼对二膦酸盐有正常反应[3]。此外,也有人将血清25-(OH)D≥30ng/ml定为正常,20~30ng/ml定为不足,可以导致骨质疏松症,但一般以骨骼外病变为主。维生素D缺乏引起的佝偻病/骨质软化症是以骨基质钙盐沉着与骨矿化障碍为主的慢性代谢性骨病,主要表现为骨组织的类骨质(未钙化的骨基质)聚积过多。病变如发生在生长中的骨骼,则成佝偻病,多见于婴幼儿,称为婴幼儿佝偻病(infant rickets)。发生在年龄较大的儿童者称为晚发性佝偻病(delayed rickets),较为少见。病变发生在骨生长发育已停止的成年人称为骨质软化症。佝偻病和骨质软化症的病因及病变特征基本相同。由于室外活动及日照减少以及城市空气污染等原因,维生素D营养性佝偻病与骨质软化症有所增多。事实上,除经典的佝偻病和骨质软化症外,骨质疏松、肌病、肥胖、高血压、免疫功能障碍和某些肿瘤亦与低维生素D状态相关[4,5]。引起维生素D缺乏/不足的原因很多,常见者见表1。
表1 维生素D缺乏/不足的常见病因

(一)维生素D与钙和磷缺乏/不足
引起维生素D、钙、磷缺乏/不足的原因很多,主要见于日光照射不足、饮食供给不足和肠道吸收障碍。
1.日光照射不足
日光的紫外线照射皮肤可形成维生素D3。由于玻璃也能吸收大部分日光中的紫外线,故室内工作者血维生素D和25-(OH)D低于室外工作者。不经常在室外活动的儿童其佝偻病的患病率要比经常在室外活动的儿童高7~8倍。热带和亚热带阳光充足,佝偻病的发生较温带和寒带少。地理位置与日照量关系密切,东北地区幼儿佝偻病的发病率明显高于华北和西北地区。长江流域佝偻病发病率高于华南。哈尔滨寒冷且日照较少,2~4月由于寒冷,儿童到室外活动少,佝偻病活动期在幼童高达43.5%,随着天气暖和情况好转,9~10月最轻,11月以后又渐加重。老年人户外活动较少且日照机会少,日照时皮肤合成活性维生素D的能力较低,易有维生素D缺乏/不足。脑瘫患者由于四肢瘫痪,日照甚少,长骨骨折发生率高,以上肢骨折常见。这与日照少及用抗癫痫药有关,服用维生素D 5000U/d,3个月治疗使临床情况显著好转。在此治疗期间血清钙与磷的平均值升高,ALP下降,因此认为维生素D不足是主要原因。
2.维生素D/钙/磷/镁供给不足
如果日照不足,就要靠食物中补充维生素D。钙、磷和镁是重要的骨矿物质。其中钙和磷尤为重要,若钙和/或磷缺乏则骨矿化不足,发生佝偻病或骨质软化症。维生素D缺乏/不足则肠道对于钙和磷吸收减少,发生钙与磷不足。发生营养不良性佝偻病/骨质软化症的主要原因是维生素D缺乏/不足,其次是缺钙,再其次是缺磷。镁是骨矿物质的重要成分,镁缺乏则甲状旁腺分泌PTH不足,且 PTH在周围组织作用欠佳,间接影响骨代谢。
3.肠道吸收障碍
血液25-(OH)D从肝脏排出后有85%被重吸收。这一肠-肝循环在肝、胆或肠有疾病时会导致维生素D缺乏/不足。胃切除或肠吸收不良综合征引起维生素D、钙、磷和镁的不足。消化系统疾病既可引起骨矿化障碍而发生佝偻病/骨质软化症,又可导致骨质疏松症。
4.其他因素
孕妇和哺乳期的维生素D需要量增加,维生素D不足引致胚胎维生素D及钙、磷等不足。婴儿于出生后所获得的母乳营养成分欠佳,故易发生婴儿佝偻病[6-8]。Klein等报道一小孩经肝活检确诊为慢性胆汁阻塞性肝病,引致肝性骨病。用维生素D治疗效果欠佳。予以紫外线照射治疗仍然不能使25-(OH)D升高。其骨钙素低,提示成骨不足。胆汁阻塞性肝病所引起的肝性骨病病因和发病机制复杂,其中包括皮肤合成维生素D减少、血液25-(OH)D下降和骨形成下降。某些药物可能引起佝偻病/骨质软化症。环孢素或tacrolimus可引起骨丢失。大剂量时引起生长板体积增大而密度降低,呈佝偻病/骨质软化症病理表现。
(二)单纯维生素D缺乏/不足
人类是一种杂食高等动物。与一般的杂食动物或草食动物比较,在长期的进化过程中,因为以下原因,人类获得维生素D的两条途径均受到限制,容易发生维生素D缺乏/不足[9]:①正常的衣着加上特殊的宗教风俗减少了皮肤与阳光的接触机会,告别猎食和农耕时代后,人类接触阳光的机会进一步减少;室外/野外活动减少和室内工作减少了阳光紫外线照射;②人类的寿命明显长于其他动物,随着增龄,阳光暴露和富含维生素D的食物摄取减少,肾脏1α-羟化酶活性降低;③高纬度老年居民、皮肤色素较深、肥胖或独居者;④冬季;⑤长年佩戴面纱或涂抹防晒霜;⑥空气污染;⑦吸烟;⑧肠吸收不良综合征;⑨慢性肝肾疾病;⑩长期使用抗惊厥药、糖皮质激素或免疫抑制剂。
Holick等对维生素D缺乏/不足的定义如下:①血清25-(OH)D 浓度<20ng/ml(50nmol/L)定为维生素 D 缺乏;②20~30ng/ml(50~75nmol/L)维生素 D 不足;③30~60ng/ml(75~150nmol/L)定为正常;④>375nmol/L(150ng/ml)定为维生素D中毒。由于25-(OH)D检测方法的准确性存在争议,加上25-(OH)D水平受多种因素的影响。因此,对维生素D缺乏/不足的定义尚不统一。从最近的50个研究结果看,全球维生素D和钙缺乏/不足的发生率高达30%~80%,老年人、较贫困地区的发病率可能更高。如果以血清25-(OH)D 30nmol/L作为维生素 D缺乏/不足的切点,50nmol/L作为维生素D充足的切点,那么全球的维生素D缺乏/不足/不足流行情况见表2~表7。
表2 成人维生素D缺乏/不足流行状况(%)
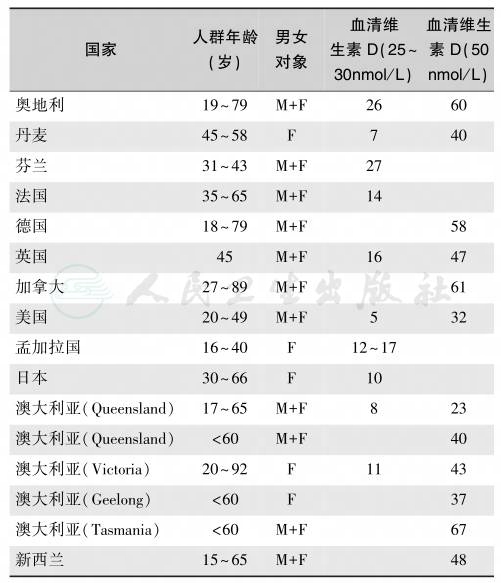
注:M:男性;F:女性;后同
表3 儿童和青少年维生素D缺乏/不足流行状况(%)
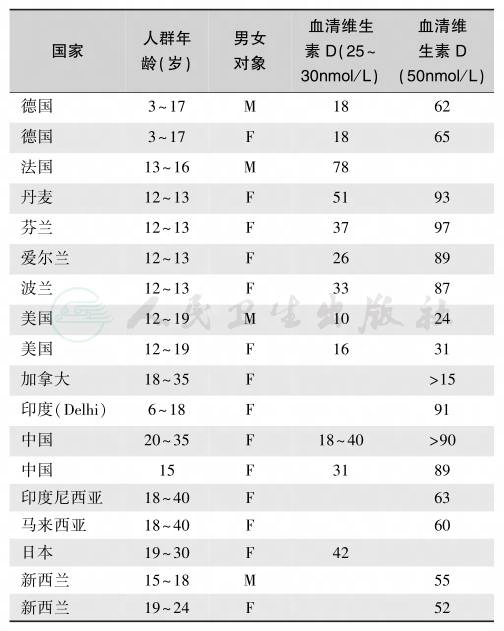
表4 老年人维生素D缺乏/不足流行状况(%)
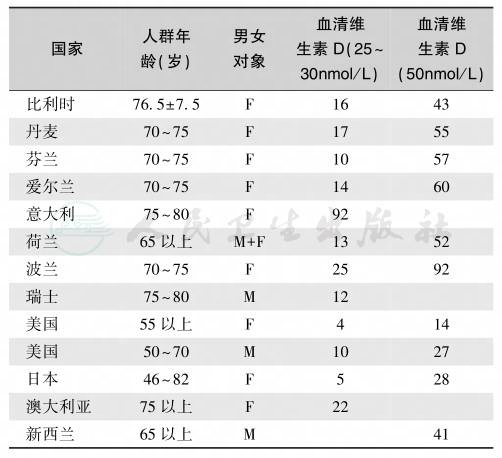
表5 成人营养性钙缺乏/不足流行状况
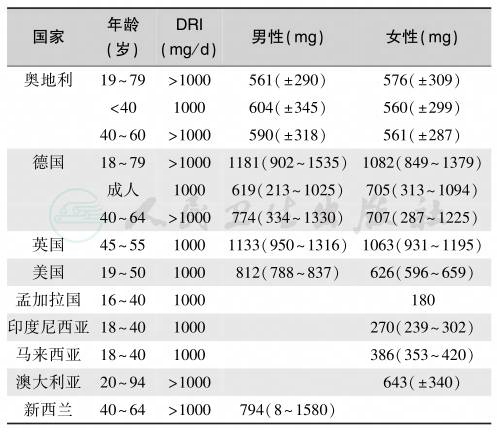
注:DRI:dietary reference intake,FAO/WHO 建议的饮食参考摄入量;中值为90%CI或95%CI
表6 老年人营养性钙缺乏/不足流行状况
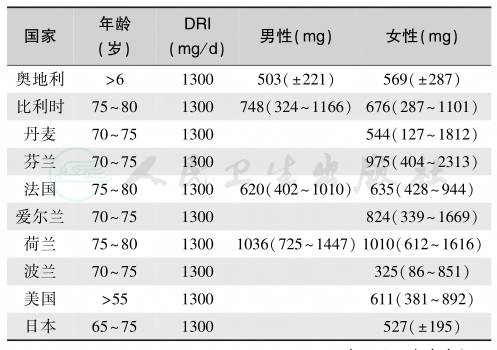
注:DRI:dietary reference intake,FAO/WHO 建议的饮食参考摄入量;中值为97.5%CI
表7 儿童与青少年营养性钙缺乏/不足流行状况
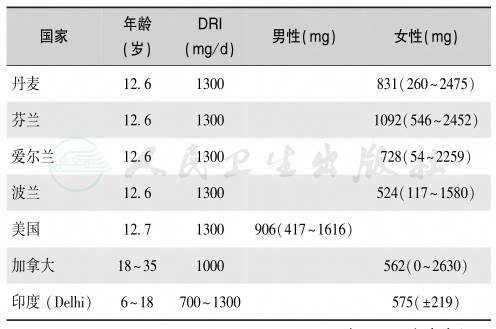
注:DRI:dietary reference intake,FAO/WHO 建议的饮食参考摄入量;中值为97.5%CI
(三)焦磷酸盐与骨关节矿化
骨矿化模型显示,焦磷酸盐(PPi)抑制羟磷灰石的形成与矿化,这一过程主要涉及三种调节蛋白。胞外液PPi是羟磷灰石成核和生长的强抑制剂,通过来源于细胞内液的ANKH(腺苷环化酶)转运和细胞外液的外核苷酸焦磷酸酶-膦酸二酯酶-1(ENPP1,由enpp1基因编码)水解ATP而补充细胞外液PPi池;另一方面,焦磷酸被组织非特异性碱性磷酸酶(TNAP)水解(图1)。对于肾脏来说,近曲小管的PPi来源于肾小球的滤过和肾集合管的焦磷酸盐细胞外液的外核苷酸焦磷酸酶-膦酸二酯酶-3(NPP3,由enpp3基因编码)水解;远曲小管分泌ATP,通过底部外侧膜的NPP1和顶部胞膜的三磷酸-二膦酸水解酶(tNTPD-2、NTPD-3)补充细胞外液的PPi池;集合管的皮质段水孔蛋白-2(AQP-2)表达ANKH,将PPi由细胞质转运至肾小管管腔及细胞间质;细胞外PPi和蛋白晶体抑制剂(如骨桥素)阻滞腔内和间质的矿盐沉着与结石形成(图2)。
(四)青春期的维生素D-PTH代谢特点
循环血液25-(OH)D反映个体的维生素D营养状态,应用此指标评价青少年的维生素D缺乏率0%~32%,差异与季节和居住纬度有关,如果将维生素D不足也算在内的化,某些地区的发病率增加到75%,见表8。
维生素D是维持正常青春期发育的必需营养素和激素,维生素D缺乏必然影响钙吸收、骨骼健康和青春期发育。
(五)老年人维生素D缺乏/不足
在美国,50%以上的老年人血清25-(OH)D低于75nmol/L,30%低于50nmol/L。老年人维生素D缺乏/不足的原因是:①暴露于阳光下,皮肤合成7-脱氢胆甾醇的能力下降(只有年轻人的25%);②阳光接触机会减少;③肥胖和体脂增加25-(OH)D分布容量,生物可用性降低;④25-(OH)D减少导致活性维生素 D生成不足;⑤肾功能下降时,1α-羟化酶活性不足,1,25-(OH)2D 合成减少;⑥IGF-1、降钙素、雌激素下降使1α-羟化酶活性减弱;⑦1,25-(OH)2D 的分解随年龄而增加。从30岁到80岁,肾小球滤过率(GFR)下降50%~63%,但由于同时期的肌肉容量亦降低,故血清肌酐仍正常;如果老年人出现血清肌酐升高,提示其GFR减低已经相当明显,仅在轻度急性应激(如手术或创伤)时即可发生尿毒症,年龄与血清肌酐、肌酐清除率的关系见图3。
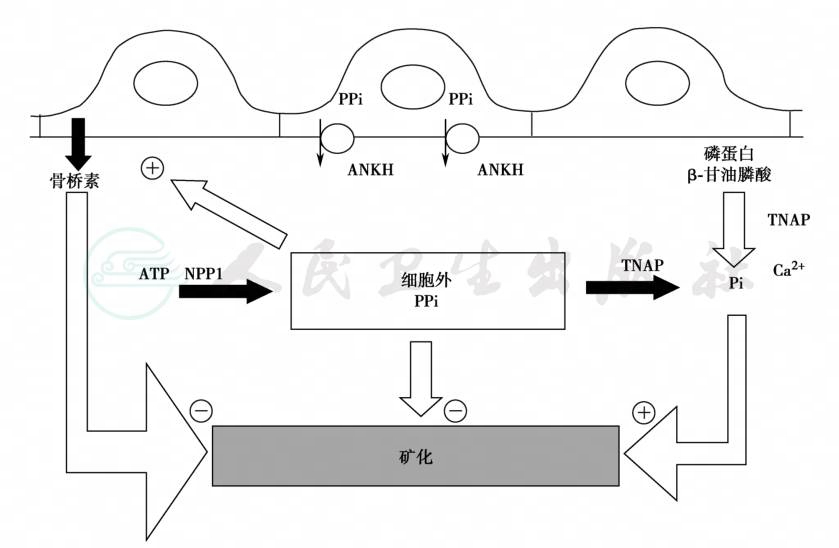
图1 成骨细胞和软骨细胞的焦磷酸盐与骨矿化
 代表抑制作用;⊕代表兴奋作用,即诱导骨桥素(osteopontin)表达并降低矿化,而无机磷(Pi)促进矿化
代表抑制作用;⊕代表兴奋作用,即诱导骨桥素(osteopontin)表达并降低矿化,而无机磷(Pi)促进矿化
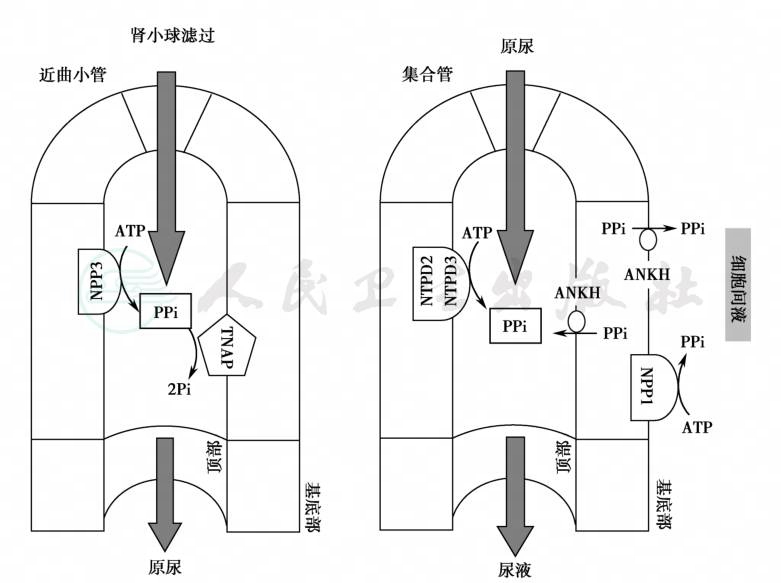
图2 肾集合管焦磷酸盐与骨矿化
表8 青春期维生素D不足/缺乏症
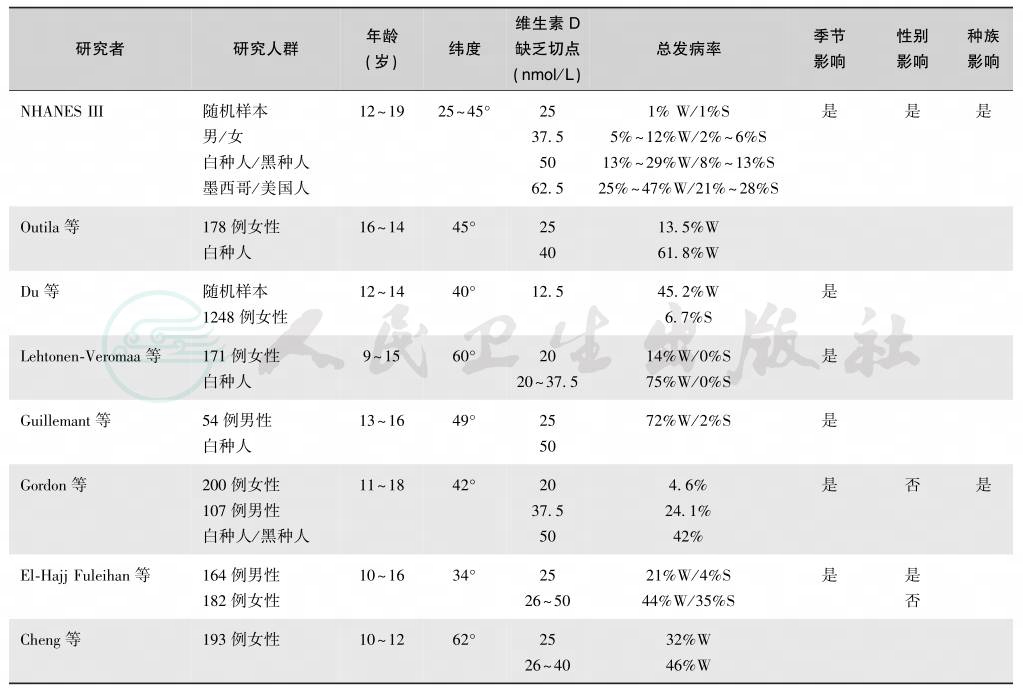
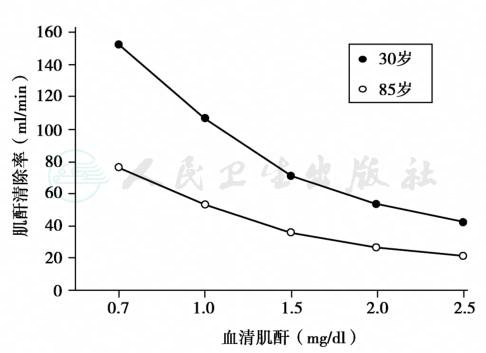
图3 年龄与血清肌酐和肌酐清除率的关系
用70kg体重标化(Cockroft-Gault公式),血清肌酐以μmol/L表示时,×88.4
1.肾脏1α-羟化酶活性不足
1α-羟化酶的活性受许多因素的调节,除PTH外,上调肾脏1α-羟化酶表达的因素还有IGF-1、降钙素和雌激素,而下调其表达的因素有血钙和血磷。上调因子水平降低或下调因子水平升高都可导致 1α-羟化酶活性不足,从而引起 1,25-(OH)2D 的合成减少。
2.肾外组织1α-羟化酶减少
维生素D受体除与维生素D反应元件结合外,还可以与 β-连环蛋白(β-catenin)结合,而后者是Wnt信号通路中的关键转录因子。因此,维生素D受体与β-连环蛋白结合后,维生素D受体可能对Wnt通路有阻滞作用,从而起到抗增殖作用。此外,1,25-(OH)2D的非基因组途径对 Wnt信号途径也有作用。肾外许多组织表达1α-羟化酶。肾外1α-羟化酶的表达受多种因素调节,但对1,25-(OH)2D不敏感。例如,成骨细胞1α-羟化酶表达不受 1,25-(OH)2D 的影响,而 IL-1β(NF-κB激动剂)兴奋其表达。在巨噬细胞中,TNF-α和IFN-γ调节1α-羟化酶活性;而在血管内皮细胞中,1α-羟化酶在PTH和雌激素的作用下,表达增加。肾外1α-羟化酶还受年龄的影响,随年龄增长而表达下降。肾外1α-羟化酶的表达情况见表9。近年发现,FGF-23和klotho调节钙磷和维生素D代谢。FGF-23抑制肾Na-Pi转运体,增加磷排泄,同时降低肾1α-羟化酶表达,上调24-羟化酶表达;在甲状旁腺,FGF-23下调PTH表达,抑制其分泌,因此FGF-23导致1,25-(OH)2D水平下降,klotho是 FGF-23的辅助蛋白,与FGF受体结合后增强了FGF-23的信号途径。总之,肾外组织表达一定量的 1α-羟化酶,并且具有产生 1,25-(OH)2D 的能力,肾外组织产生的1,25-(OH)2D也可能有少量进入血液循环参与钙磷代谢的调节,其中甲状旁腺组织产生的1,25-(OH)2D还对局部PTH的合成有抑制作用,因此,老年人肾外组织1,25-(OH)2D的合成减少也可能在老年性骨质疏松的发病过程中起一定作用。肾脏和肾外1,25-(OH)2D的功能见图4。
表9 肾外1α-羟化酶及其调节因子

注:↓:下降;↑:升高;N:正常;+:阳性;-:阴性;?:不明
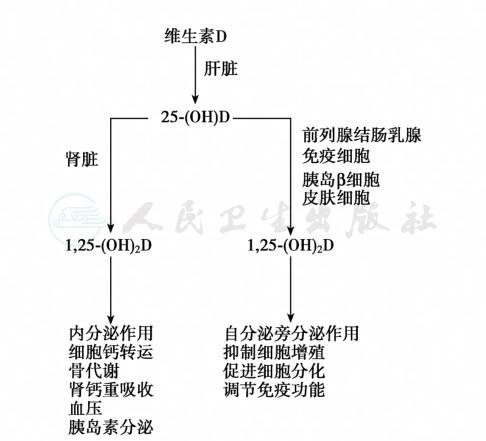
图4 肾脏和肾外1,25-(OH)2D的功能
1,25-(OH)2D 是皮肤、肌肉、骨骼、甲状旁腺和免疫系统细胞分化与增殖的调节因子,因而能影响机体组织的细胞行为,与皮肤角质细胞分化、胰岛素分泌、血压调节和免疫反应有密切关系,也与上皮细胞癌、多发性硬化、肌肉无力、骨骼疾病相关[10,11]。有些健康老年人单纯维生素 D缺乏/不足时,由于骨组织对PTH的反应差,不发生甲旁亢,仅表现为功能性甲旁减,使这些老年人的死亡率升高。
3.钙吸收减少
钙的吸收机制涉及跨细胞途径和旁细胞途经两个方面。跨细胞途径位于细胞顶端,通过细胞间的紧密连接(tight junction),由Ca2+通道将细胞外液Ca2+扩散进入细胞内;而在细胞的基底部,Ca2+泵(Na+/Ca2+交换体,Na+/Ca2+exchanger)将Ca2+主动吸收。Ca2+通道为瞬时电势受体(transient receptor potential,TRP)超家族成员,属于香草精(vanilloid subfamily,TRPV)样受体。 肾脏 Ca2+通道以 TRPV5为主,而肠道的Ca2+通道以TRPV6为主。肠钙吸收随年龄而下降,主要是因为肠道TRPV6表达下降所致。而TRPV6的表达受1,25-(OH)2D、雌激素、PTH和饮食钙摄入等因素的调节。其中,雌激素缺乏/不足和维生素D抵抗可能是老年女性肠钙吸收下降更多的原因。肾脏的TRPV5表达随年龄而下降,TRPV5主要受 1,25-(OH)2D、PTH 和 klotho的调节,其中klotho的表达下降可能是引起TRPV5表达减少的直接原因。肠钙吸收(TRPV6表达)和肾钙重吸收(TRPV5表达)通过血清Ca2+偶联。在特发性高钙尿症患者中,钙的重吸收减少引起高钙尿症,并进一步导致1,25-(OH)2D代偿性升高,使肠钙吸收增多。钙结合蛋白(calbindin)是细胞质中的钙结合因子,分为9K和25K两种亚型。9K的钙结合蛋白主要与TRPV6共存于小肠黏膜上皮细胞中,而28K的钙结合蛋白主要与TRPV5共存于肾小管上皮中。老年人或维生素D缺乏/不足时,肾和肠的钙结合蛋白表达减少,导致肠钙的主动吸收和肾的重吸收钙减少。长期应用他汀类药物的患者亦可发生维生素D缺乏/不足,而补充适量维生素D可增强他汀类药物的疗效,降低肌病发生率[12]。
(六)营养性佝偻病
营养性佝偻病(nutritional rickets,NR)仍然是发展中国家和地区生长发育期的常见健康问题。NR的病因可能是单纯维生素D缺乏症(isolated vitamin D deficiency,IVDD)、单纯钙缺乏症(isolated calcium deficiency,ICD)或两种并存,临床上以IVDD常见。除了对骨骼产生急性和慢性损害外,婴幼儿期的维生素D缺乏症(vitamin D deficiency,VDD)还可诱发1型糖尿病、肿瘤和多发性硬化症(multiple sclerosis)。VDD是NR的主要病因,其次为骨骺融合前的饮食钙缺乏/不足,据报道,土耳其的NR病因以VDD为主,而埃及和尼日利亚的NR是钙与维生素D同时缺乏/不足所致。维生素 D充足或血清25-(OH)D在20ng/ml(50nmol/L)以上者,迅速生长期的肠钙吸收可高达80%,但在维生素D缺乏/不足时,吸收率可降至10%~15%,肾脏最大磷的重吸收率亦降低,PTH分泌增多,加重磷和钙的丢失,骨矿化不良。低磷血症使肥厚性软骨细胞凋亡障碍,细胞空泡化(ballooning),生长板结构紊乱。在胎儿发育期,前软骨细胞诱导骨组织发育,生长板软骨内骨化的间质细胞积聚,继而生成成软骨细胞、软骨细胞和软骨基质。
生长板软骨细胞进行序列分化,静止带、增殖带、肥厚带和骨化带的区带分明。肥厚软骨细胞的基质骨化并在凋亡前形成初级骨化中心,血管、破骨细胞和成骨细胞侵入钙化组织,并进行组织构塑,形成次级骨化中心,骨骼生长伸长,直至骨骺融合。佝偻病时,由于低磷血症等原因,肥厚性软骨细胞的凋亡减少,使生长板软骨组织扩张,畸形且边缘不规则,在X线片上,骺端出现杯口刷状外观;严重维生素D缺乏/不足导致佝偻病骨骼病变和畸形。
胎儿期和围产期的维生素D来源是胎盘供应、母乳喂养和光照;2月龄内,其维生素D水平与母亲的血清维生素D相关,在以后的数年内主要与喂养和光照相关。因而,母亲的维生素D储存不足和喂养方式不当成为婴幼儿维生素D缺乏/不足的主要病因(表10)。
表10 维生素D营养状态与临床表现的关系
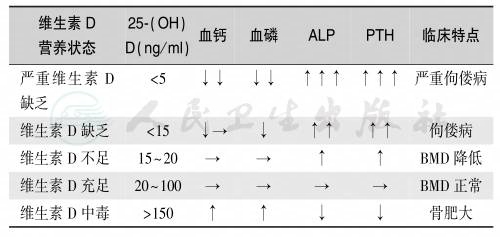
由于营养素异常所致的骨代谢紊乱称为营养性骨病,营养性骨病是骨质疏松的危险因素之一,其中维生素D缺乏/不足引起骨质软化/佝偻病和钙与维生素D不足所致的骨质疏松最常见。
(七)内源性维生素D受体缺乏
内源性维生素D受体缺乏症的病因为维生素D受体缺乏(受体表达不足,数目减少),患者表现为严重的低钙血症和高PTH血症。血清1,25-(OH)2D正常(据此可排除 1b型假性甲旁减),普通维生素 D 治疗无效而补充低剂量 1,25-(OH)2D(0.5μg/d)有效。引起血钙降低和PTH升高的病因主要有维生素D缺乏症、维生素D抵抗综合征和假性甲旁亢,因此,必须在排除这些疾病后,才能诊断内源性维生素D受体缺乏症。
增加日照、谷类和富含维生素D与钙剂食物摄入是防治维生素D缺乏/不足的经济有效方法。如果缺少阳光,则按生理维持量补充,与或不与钙剂同时服用,1周的总剂量可以一次给予。如果维生素D不足或缺乏明显,应先给予维生素D负荷量,1个月后血25-(OH)D浓度可达到高峰;但是孕妇只给予维持剂量。如果患者合并有骨质疏松症,维生素D与钙剂合用作为基础治疗。必须注意,活性维生素D或其衍生物的安全范围窄,1,25-(OH)2D与钙同时服可迅速升高血钙,因而只在1α-羟化酶缺陷症或1,25-(OH)2D生成不足时使用(如 CKD-MBD,VDDR-I,TIO,GIOP,老年人等),活性维生素D或其衍生物不能纠正维生素D缺乏症。
(一)日照
1.日光紫外线照射
日光紫外线照射虽安全,但其疗效有限,一般仅用于轻度和可疑患者的预防。一般认为,非保护性头部和双侧上臂阳光暴露每次10分钟,每周3次可达到预防维生素D缺乏/不足的目的。户外活动/钙/维生素D是预防营养不良性佝偻病的主要措施。一定的日照可改善营养状态,适当活动增进健康。孕妇与乳母有足够的营养及维生素D和钙磷,对于预防母子佝偻病和骨质软化症是重要的。日光紫外线照射能使皮下生成维生素D。一般情况下,人体10%的皮肤直接接触阳光10分钟,皮肤可合成维生素D31000U,因而,多晒太阳是补充维生素D的最经济有效措施。年龄和防晒霜是阳光照射后,皮肤合成维生素D的主要影响因素,不同季节的阳光照射的维生素D合成效果也有一定差别;皮肤色素深和老年人的效果要差一些,需要适当延长日照时间[49];涂抹防晒霜会阻挡紫外线对皮肤的照射,使皮肤不能有效地合成维生素D(图14~图16)。已经有维生素D缺乏/不足者单纯日照不能纠正缺乏/不足状态,必须口服补充。另一方面,过度接触强烈日照可引起皮肤损伤甚至诱发皮肤癌或红斑狼疮,应予避免[50]。
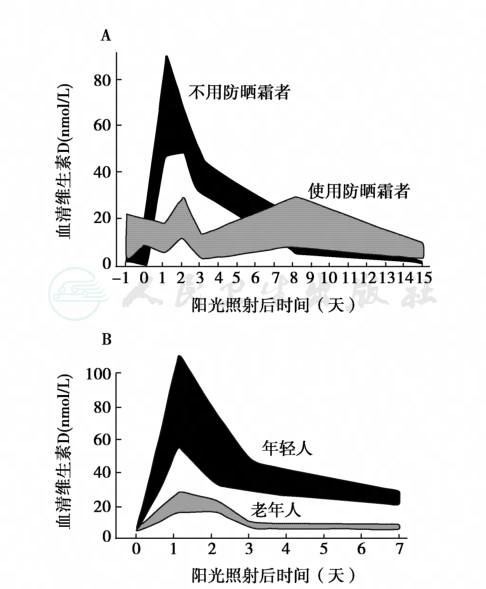
图14 单次阳光照射对血清维生素D的影响
A.单次阳光照射后,使用和不使用防晒霜对血清维生素D的影响;B.年轻人和老年人全身阳光照射对循环血维生素D浓度的影响
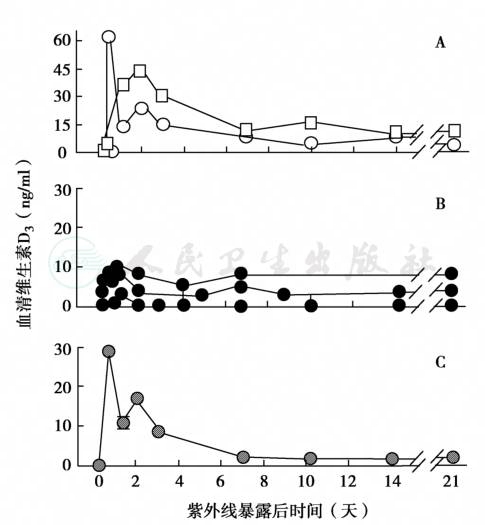
图15 皮肤色素对血清25(OH)D的影响
A、B.皮肤轻度色素沉着白种人(A,2例)与重度色素沉着黑种人(B,3例)在全身暴露紫外线照射(54mJ/cm2)后,血清 25-(OH)D浓度的差异;C.1例黑种人再次紫外线照射320mJ/cm2后的血清25-(OH)D浓度变化,均有明显差异;资料来源于The Lancet,1982,1:74-76.
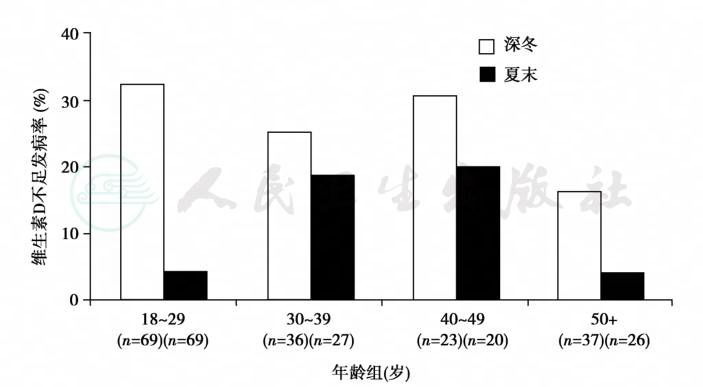
图16 年龄和季节与维生素D缺乏发生率的关系
不同年龄的人群在冬末和夏末血清25-(OH)D浓度的变化有明显差异
2.人工紫外线照射
人工紫外线照射曾用于治疗维生素D严重缺乏患者,对于行动不便或无法接触阳光照射的患者来说,人工紫外线照射预防维生素D缺乏有效。但人工紫外线照射可损伤细胞的DNA,是一种致癌原,尤其与黑色素瘤、基底细胞癌、鳞状上皮癌和其他非黑色素瘤皮肤肿瘤有密切关联。此外,紫外线照射也容易引起皮肤红斑,诱发红斑狼疮,故不主张使用[51]。
(二)食物补充预防维生素D缺乏/不足
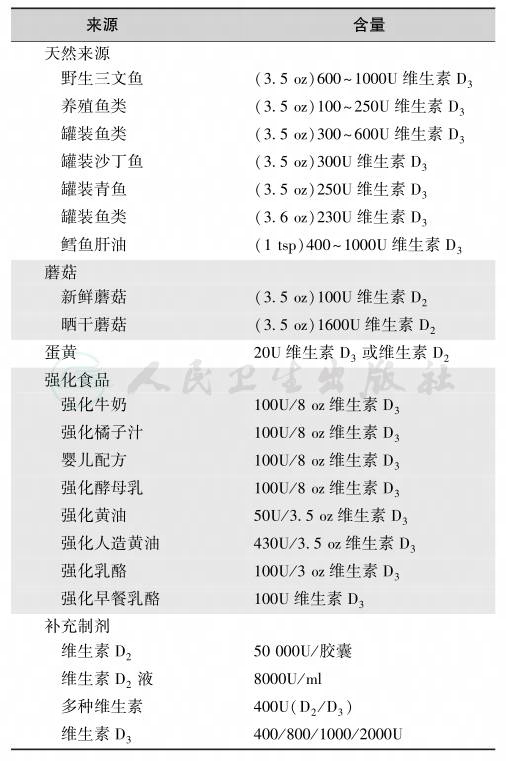
通过食物补充维生素D是预防维生素D缺乏/不足与成人骨质软化及骨质疏松的主要方法,应特别注意进食富含维生素D的食物(如鱼类、蘑菇及维生素D强化食品等),饮食维生素D、替代和药物制剂的含量见表21。
表21 饮食和药物制剂的维生素D含量
(三)维生素D治疗对象
维生素D的补充剂量与方法主要根据病因决定。预防和治疗维生素D不足/缺乏/不足症的常见情况如下:
1.维生素D不足/缺乏症
中国营养学会制定的《中国居民膳食营养素摄入量》推荐维生素D的推荐摄入量(RNI)和可耐受最高摄入量(UL)应由20μg/d提高到50μg/d,其理由:①美国食品营养委员会(FNB,1997)和欧共体食品科学委员会(SCF,2002年)的 UL均为 50μg/d;②美国营养责任委员会(CRN,2006)的UL为25μg/d。一般补充维生素D2 400~800U/d,必要时可达到2000U/d,但不能超过4000U/d;同时增加钙的摄入量或补充适量钙剂。如果患者存在维生素D吸收不良(如慢性腹泻、严重炎症性肠病、慢性胰腺炎等),应增加维生素D的补充量,如维生素D21250~5000U/d或12 500~25 000U/月;必要时亦可肌肉注射给药。
2.维生素D抵抗性佝偻病
使用普通维生素D难以达到治疗目的,一般需要使用较大剂量的活性维生素D,如1型维生素D抵抗性佝偻病(1α-羟化酶缺陷症)患者需要1,25-(OH)2D 0.5~1.0μg/d,或维生素 D22 万~10 万 U/d。同时增加钙和磷的摄入量。以前,Ⅱ型维生素D抵抗性佝偻病(维生素D受体突变)用大剂量维生素D2治疗,用量为4万~20万U/d;目前主要用 1,25-(OH)2D 治疗,剂量依个体的具体情况而定。
3.骨质疏松
对于老年性骨质疏松,在维持正常的血清25-(OH)D 的基础上,适当给予 1,25-(OH)2D 或 1α-维生素D能起一定的治疗作用。
4.其他治疗
如果临床治疗的目的主要是调节免疫功能,宜首选普通维生素D制剂。当血清25-(OH)D降低时,亦宜首选普通维生素D制剂,使其水平迅速升至正常。而在血清25-(OH)D正常时,尤其是肾脏功能严重受损时,宜首选 1,25-(OH)2D。
(四)维生素D制剂选择
1.维生素D制剂的选择
一般根据病因选择维生素D制剂,佝偻病的临床类型与维生素D制剂的选择见表22[52]。
表22 佝偻病的临床类型与处理
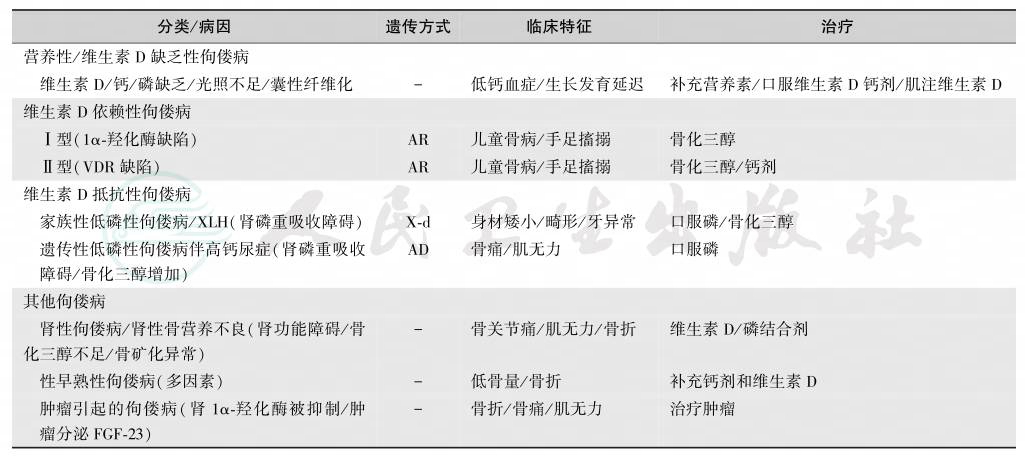
注:AD:常染色体显性遗传;AR:常染色体隐性遗传;X-d:性连锁显性
目前临床上主要有三种维生素D制剂,即普通维生素D、1α-维生素 D 和1,25-(OH)2D(表23)。 临床上应根据病情需要和各种维生素D制剂的优缺点进行合理选择。
表23 三种维生素D制剂的主要区别

2.维生素D 2和维生素D 3的效应差别
以前认为,维生素D2和维生素D3的效价相等,并且可以互相换算。但是,近年的资料表明,维生素D2和维生素D3的作用强度、作用时间和使用途径均有差异,应该引起足够重视。事实上,当以血清25-(OH)D水平作为维生素D营养状态评价指标时,维生素D2维持血清25-(OH)D正常水平的时间与维生素D受体结合亲和力均明显低于维生素D3,一般前者为后者的1/2左右。荟萃分析有关的RCT研究发现,补充D3后升高血清25-(OH)D的幅度明显高于同等剂量的D2[53]。
3.口服与肌注的效应差别
一般口服维生素D较肌注维生素D的达峰时间明显增快,口服达峰时间约数天,而肌注的达峰时间需要1个月以上。其次,口服维生素D较肌注更接近生理状况;因而一般仅在存在肠吸收不良时,考虑由注射途径补充维生素D。第三,肌注补充的维生素D难以进入外周组织,而口服维生素D可以通过正常途径,在外周组织1α-羟化酶作用下生成1,25-(OH)2D;也就是说,口服途径同时纠正了外周组织的维生素D缺乏/不足状态,这对于维持组织中1,25-(OH)2D的正常水平十分重要。研究发现,给健康志愿者单次肌注600 000U的胆钙化醇(cholecalciferol,维生素 D3)仅使35%的对象达到理想(充足)血清 25-(OH)D水平,25%仍降低,而PTH的变化不明显。所以,单次高剂量注射不能完全纠正维生素D缺乏/不足状态[54]。
4.维生素D和活性维生素D的效应差别
一般来讲,对于肾功能正常者来说,没有必要应用活性维生素D,而补充足够量的维生素D即可达到治疗目的。而且补充维生素D可能还较活性维生素D有更多优点,例如作用时间长,费用低廉等。但是,对于肾功能减退的患者来说,加用活性维生素D可能具有更好的效果。
5.维生素D个体差别
维生素D是决定血钙水平的最重要因素,因而血清的维生素D与血钙水平存在相关关系。一般来说,每补充100U维生素D3大约升高血25-(OH)D 1ng/ml;如果患者的血清25-(OH)D为20ng/ml,欲将其水平升高至30ng/ml的维生素 D3补充量大约是:100×(30-20)=1000U。 但是,因为血钙和体内的维生素D代谢与活性还受年龄、肾功能、肝功能、基础疾病与钙摄入量等因素的影响,因而,个体的差异性大,补充维生素D期间必须定期监测血钙、尿钙和血清25-(OH)D。
6.年龄差别
儿童(尤其是新生儿)患者应使用专门的维生素D制剂,这些制剂包括:①维生素D滴剂(400U/ml);②维生素D胶囊(400U/ml);③多种维生素D胶囊(含维生素D 400U/ml和其他维生素)。老年患者因肾功能降低及平衡能力下降,应主要考虑补充活性维生素D。在大多数情况下,如果血 25-(OH)D<20ng/ml,每周可给予 D2或 D3 5000U,共8周,然后用以下三种方法之一进行维持治疗:①每2周给予D25000U;②每天给予维生素D 1000~2000U;③阳光照射。3~6个月后再行复查,视情况停止或继续治疗。老年人是发生维生素D不足或缺乏的高危人群,而且维生素D不足或缺乏与2型糖尿病、心血管病、高血压、血脂谱异常、哮喘、感染、骨质疏松等有关。每天给予维生素D3 2500U,共治疗28天,可使50%以上的儿童维生素D缺乏症[血清25-(OH)D低于30ng/ml]患者得到纠正,但其中半数的血清25-(OH)D会逐渐下降;如果需要,可继续使用6~18个月,但必须定期监测血清25-(OH)D水平[22]。多发性硬化的发病主要与紫外线照射、维生素D和维生素D受体的多态性有关,大量的资料表明,多发性硬化主要发生于高纬度的地区,而经常接触阳光紫外线或补充维生素D可以降低多发性硬化的发病率。
理想的维生素D水平是骨骼健康和骨质疏松症防治的基本要求,维生素D被列为骨质疏松症防治的基础药物和重要营养素。理想的25-(OH)D水平应满足以下要求:①能最大限度地抑制血PTH浓度;②能达到最大的钙吸收;③能达到最高的骨密度;④能最大限度地降低骨丢失率。⑤能最大限度地降低骨折率。多数学者认为,能预防骨折的血清25-(OH)D 水平估计为 75~80nmol/L(30~32ng/ml)。 根据 IOF的建议,补充维生素D的剂量取决于个体用药前的25-(OH)D水平、BMI和有效日照。降低跌倒风险 20%的剂量为17.5~25μg/d,降低非椎体和髋部骨折风险20%的剂量为10~20μg/d。需补充的剂量可根据测量值估算,补充每1μg的维生素D,可增加血清 25-(OH)D 1ng/ml。 NOF推荐,50岁以上者的剂量为800~1000U/d(20~25μg/d)。服用维生素D应定期监测血钙、钙磷乘积及PTH水平,并根据监测结果调整剂量[55-57]。
(五)钙剂和维生素D治疗方案
一般来说,白种人、集体居住老年人每天的维生素D需要量为10~20μg(400~800U),使血 25-(OH)D 达到 20~30ng/ml(50~75nmol/L),体弱多病或住院患者每天可能需要 50μg(2000U),补充的方法以每日口服为佳。但必须监测25-(OH)D水平,防止发生维生素D中毒。除高钙血症外,维生素D过量同样引起骨质疏松症。大剂量维生素D用于佝偻病治疗,严重患者可肌注维生素D3(胆钙化醇)每次30万U,必要时2~4周重复1次。三种营养素的补充见表24。在维生素D的补充治疗中,应注意这些问题。
表24 营养性佝偻病与骨质软化症的原因与防治
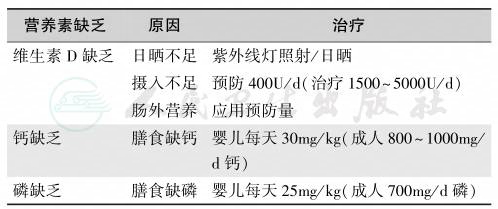
注:维生素D对于佝偻病用治疗量,治愈后用预防量
1.Viswanathan HN,Curtis JR,Yu J,et al.Direct healthcare costs of osteoporosis-related fractures in managed care patients receiving pharmacological osteoporosis therapy.Appl Health Econ Health Policy,2012,10(3):163-173.
2.Kurtinaitis J,Dadonien J,Kvederas G,et al.Mortality after femoral neck fractures:a two-year follow-up.Medicina (Kaunas),2012,48(3):145-149.
3.Emohare O,Wiggin M,Hemmati P,et al.Assessing Bone Mineral Density Following Acute Hip Fractures:The Role of Computed Tomography Attenuation.Geriatr Orthop Surg Rehabil,2015,6(1):16-21.
4.林华.骨质疏松性骨折及其影响愈合因素.中国实用内科杂志,2011,31(7):236-238.
5.Kim DH,Vaccaro AR.Osteoporotic compression fractures of the spine:current options and considerations for treatment.Spine J,2006,6(5):479-487.
6.Gehlbach SH,Avrunin JS,Puleo E,et al.Fracture risk and antiresorptive medication use in older women in the USA.Osteoporos Int,2007,18:805-881.
7.Kanis JA,Johnell O,Oden A.Ten year probabilities of osteoporotic fractures according to BMD and diagnostic thresholds.Osteoporos Int,2001,12:989-995.
8.Fox K,Magaziner J,Hawkes W,et al.Loss of bone density and lean body mass after hip fracture.Osteoporos Int,2000,11(1):31-35.
9.Delmas PD.The use of biochemical markers of bone turnover in the management of post-menopausal osteoporosis.Osteoporos Int,2000,11(1):S1-S6.
10.林华.骨质疏松性骨折的危险因素.国际内分泌代谢杂志,2006,26(4):236-238.
11.Rizzoli R,Bruyere O,Cannata-Andia JB,et al.Management of osteoporosis in the elderly.Curr Med Res Opin,2009,25(10):2373-2387.
12.Morosano M,Masoni A,Sanchez A.Incidence of hip fractures in the city of Rosario,Argentina.Osteoporos Int,2005,16(11):1339-1344.
13.van Schoor NM,Smit JH,Bouter LM et al.Maximum potential preventive effect of hip protectors.J Am Geriatr Soc,2007,55(4):507-510.
14.林华,徐天舒,范璐等.唑来膦酸盐(5mg)干预绝经后骨质疏松症对骨量的影响.中华骨科杂志,2011,31(12):1331-1336.
15.Nandi SK,Kundu B,Ghosh SK,et al.Efficacy of nano-hydroxyapatite prepared by an aqueous solution combustion technique in healing bone defects of goat.J Vet Sci,2008,9(2):183.
16.Honig S.Osteoporosis New Treatments and Updates.Bull NYU Hosp Jt Dis,2010,68(3):166-170.
17.Anderson GL,Limacher M,Assaf AR,et al.Effects of conjugated equine estrogen in postmenopausal women with hysterectomy:the Women’s Health Initiative randomized controlled trial.J Am Med Assoc,2004,291(14):1701-1712.
18.Pereira RM,Carvalho JF,Paula AP,et al.Guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis.Rev Bras Reumatol,2012,52(4):580-593.
19.McCloskey E,Kanis JA.FRAX updates 2012.Curr Opin Rheumatol,2012,24(5):554-560.
20.Poku EK,Towler MR,Cummins NM,et al.Developing novel prognostic biomarkers for multivariate fracture risk prediction algorithms.Calcif Tissue Int,2012,91(3):204-214.
21.Hopkins RB,Goeree R,Pullenayegum E,et al.The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in postmenopausal women.BMC Musculoskeletal Disorders,2011,12:209.
22.Leslie WD,Lix LM,Langsetmo L,et al.Construction of a FRAX®model for the assessment of fracture probability in Canada and implications for treatment.Osteoporos Int,2011,22:817-827.
23.Fraser LA,Langsetmo L,Berger C,et al.Fracture prediction and calibration of a Canadian FRAX® tool:a population-based report from CaMos.Osteoporos Int,2011,22(3):829-837.
24.Kanis JA,McCloskey E,Johansson H,et al.FRAX with and without Bone Mineral Density.Calcif Tissue Int,2012,90(1):1-13.
25.Arden NK,Griffiths GO,Hart DJ,et al.The association between osteoarthritis and osteoporotic fracture:the Chingford Study.Br J Rheumatol,1996,35:1299-1304.
26.Astrom J,Beertema J.Reduced risk of hip fracture in the mothers of patients with osteoarthritis of the hip.J Bone Joint Surg Br,1992,74(2):270-271.
27.Biyani A,Simison AJ,Klenerman L.Intertrochanteric fractures of the femur and osteoarthritis of the ipsilateral hip.Acta Orthop Belg,1995,61(2):83-91.
28.Li X,Wang XQ,Chen BL,et al.Whole-Body Vibration Exercise for Knee Osteoarthritis:A Systematic Review and Meta-Analysis.Evid Based Complement Alternat Med,2015,2015:758147.
29.Cumming RG,Klineberg RJ.Epidemiological study of the relation between arthritis of the hip and hip fractures.Annals of the rheumatic diseases,1993,52:707-710.
30.Dequeker J,Johnell O.Osteoarthritis protects against femoral neck fracture:the MEDOS study experience.Bone,1993,14(1):S51-56.
31.Foss MV,Byers PD.Bone density,osteoarthrosis of the hip,and fracture of the upper end of the femur.Ann Rheum Dis,1972,31(4):259-264.
32.Arden NK,Nevitt MC,Lane NE,et al.Osteoarthritis and risk of falls,rates of bone loss,and osteoporotic fractures.Study of Osteoporotic Fractures Research Group.Arthritis Rheum,1999,42:1378-1385.
33.Jones G,Nguyen T,Sambrook PN,et al.Osteoarthritis,bone density,postural stability,and osteoporotic fractures:a population based study.J Rheumatol,1995,22:921-925.
34.Tam HH,Bhaludin B,Rahman F,et al.SPECT-CT in total hip arthroplasty.Clin Radiol,2014,69(1):82-95.
35.Wand JS,Hill ID,Reeve J.Coxarthrosis and femoral neck fracture.Clin Orthop Relat Res,1992,(278):88-94.
36.Antoniades L,MacGregor AJ,Matson M,et al.A cotwin control study of the relationship between hip osteoarthritis and bone mineral density.Arthritis Rheum,2000,43(7):1450-1455.
37.Styrkarsdottir U,Halldorsson BV,Gretarsdottir S,et al.Multiple genetic loci for bone mineral density and fractures.N Engl J Med,2008,358(22):2355-2365.
38.Hilliquin P,Pessis E,Coste section sign J,et al.Quantitative assessment of joint space width with an electronic caliper.Osteoarthritis Cartilage,2002,10:542-546.
39.Kellgren JH,Lawrence JS.Radiological assessment of osteo-arthrosis.Annals of the rheumatic diseases,1957,16(4):494-502.
40.Emrani PS,Katz JN,Kessler CL,et al.Joint space narrowing and Kellgren-Lawrence progression in knee osteoarthritis:an analytic literature synthesis.Osteoarthritis Cartilage,2008,16(8):873-882.
41.Schiphof D,Boers M,Bierma-Zeinstra SM.Differences in descriptions of Kellgren and Lawrence grades of knee osteoarthritis.Ann Rheum Dis,2008,67(7):1034-1036.
42.Hoch JM,Mattacola CG,Medina McKeon JM,et al.Serum cartilage oligomeric matrix protein(sCOMP)is elevated in patients with knee osteoarthritis:a systematic review and meta-analysis.Osteoarthritis Cartilage,2011,19(12):1396-1404.
43.Raja K,Dewan N.Efficacy of knee braces and foot orthoses in conservative management of knee osteoarthritis:a systematic review.Am J Phys Med Rehabil,2011,90(3):247-262.
44.Mosher TJ,Zhang Z,Reddy R,et al.Knee articular cartilage damage in osteoarthritis:analysis of MR image biomarker reproducibility in ACRIN-PA 4001 multicenter trial.Radiology,2011,258(3):832-842.
45.Wuermser LA,Achenbach SJ,Amin S,et al.What accounts for rib fractures in older adults? J Osteoporos,2011;2011:457591.
46.Palvanen M,Kannus P,Niemi S,et al.Hospital-treated minimal-trauma rib fractures in elderly Finns:long-term trends and projections for the future.Osteoporosis International,2004,15(8):649-653.
47.Barrett-Connor E,Nielson CM,Orwoll E,et al.Epidemiology of rib fractures in older men:Osteoporotic Fractures in Men (MrOS)prospective cohort study.British Medical Journal,2010,340:p.c1069.
48.González-Reimers E,Quintero-Platt G,Rodríguez-Rodríguez E,et al.Bone changes in alcoholic liver disease.World J Hepatol, 2015, 7(9):1258-1264.
49.Raisz LG.Clinical practice.Screening for osteoporosis.N Engl J Med,2005,353(2):164-171.
50.Miller PD,Siris ES,Barrett-Connor E,et al.Prediction of fracture risk in postmenopausal white women with peripheral bone densitometry:Evidence from the National Osteoporosis Risk Assessment.J Bone Miner Res,2002,17(12):2222-2230.
51.Stone KL,Seeley DG,Lui LY,et al.BMD at multiple sites and risk of fracture of multiple types:long-term results from the Study of Osteoporotic Fractures.J Bone Miner Res,2003,18(11):1947-1954.
52.Barnea Y,Kashtan H,Skornick Y,et al.Isolated rib fractures in elderly patients: mortality and morbidity.Canadian Journal of Surgery,2002,45(1):43-46.
53.Papaioannou A,Kennedy CC,Ioannidis G,et al.The impact of incident fractures on health-related quality of life:5 years of data from the Canadian Multicentre Osteoporosis Study.Osteoporos Int, 2009, 20(5):703-714.
54.Melton LJ,3rd,Gabriel SE,Crowson CS,et al.Cost-equivalence of different osteoporotic fractures.Osteoporos Int,2003,14(5):383-388.
55.Melton LJ,3rd,Crowson CS,O’Fallon WM.Fracture incidence in Olmsted County,Minnesota:comparison of urban with rural rates and changes in urban rates over time.Osteoporos Int,1999,9(1):29-37.
56.Sanders KM,Seeman E,Ugoni AM,et al.Age-and gender-specific rate of fractures in Australia: a population-based study.Osteoporos Int,1999,10(3):240-247.
57.Cooley H,Jones G.A population-based study of fracture incidence in-Southern Tasmania:lifetime fracture risk and evidence for geographic variations within the same country.Osteoporos Int,2001,12(2):124-130.









