


 收藏
收藏 已收藏
已收藏英文名称 :succinic semialdehyde dehydrogenase deficiency
中文别名 :4-羟丁酸尿症;γ-羟丁酸血症
琥珀酸半醛脱氢酶缺乏症(succinic semialdehyde dehydrogenase deficiency,SSADH)也称为 4-羟基丁酸尿 症(4-hydroxybutyric aciduria),由 Jakobs于 1981 年首次报道,为神经递质γ-氨基丁酸(γ-aminobutyric acid,GABA)代谢异常性疾病。
琥珀酸半醛脱氢酶缺陷病是由于琥珀酸半醛脱氢酶缺陷导致的一种罕见的遗传性代谢病,是儿童γ-氨基丁酸(γ-aminobutyric acid,GABA)代谢疾病常见的一种,40%的病例存在父母近亲婚配现象。1981年由Jakobs首次报告,为一种罕见的常染色体隐性遗传病,自1981年首次报道至2011年全世界报道逾450例。
为常染色体隐性遗传性疾病,致病基因ALDH5A1位于6p22,编码琥珀酸半醛脱氢酶,该酶参与了γ-氨基丁酸的代谢过程(图1),首先γ-氨基丁酸在γ-氨基丁酸转氨酶的作用下,形成琥珀酸半醛,然后在琥珀酸半醛脱氢酶的作用下形成琥珀酸,参与三羧酸循环。琥珀酸半醛脱氢酶缺乏时,琥珀酸半醛经旁路形成4-羟基丁酸,血、尿、脑脊液中4-羟基丁酸显著增多,故本病又称4-羟基丁酸尿症。过量的4-羟基丁酸对神经系统产生毒性作用,而神经递质γ-氨基丁酸的代谢异常及线粒体功能异常也参与了琥珀酸半醛脱氢酶缺乏症的病理过程。
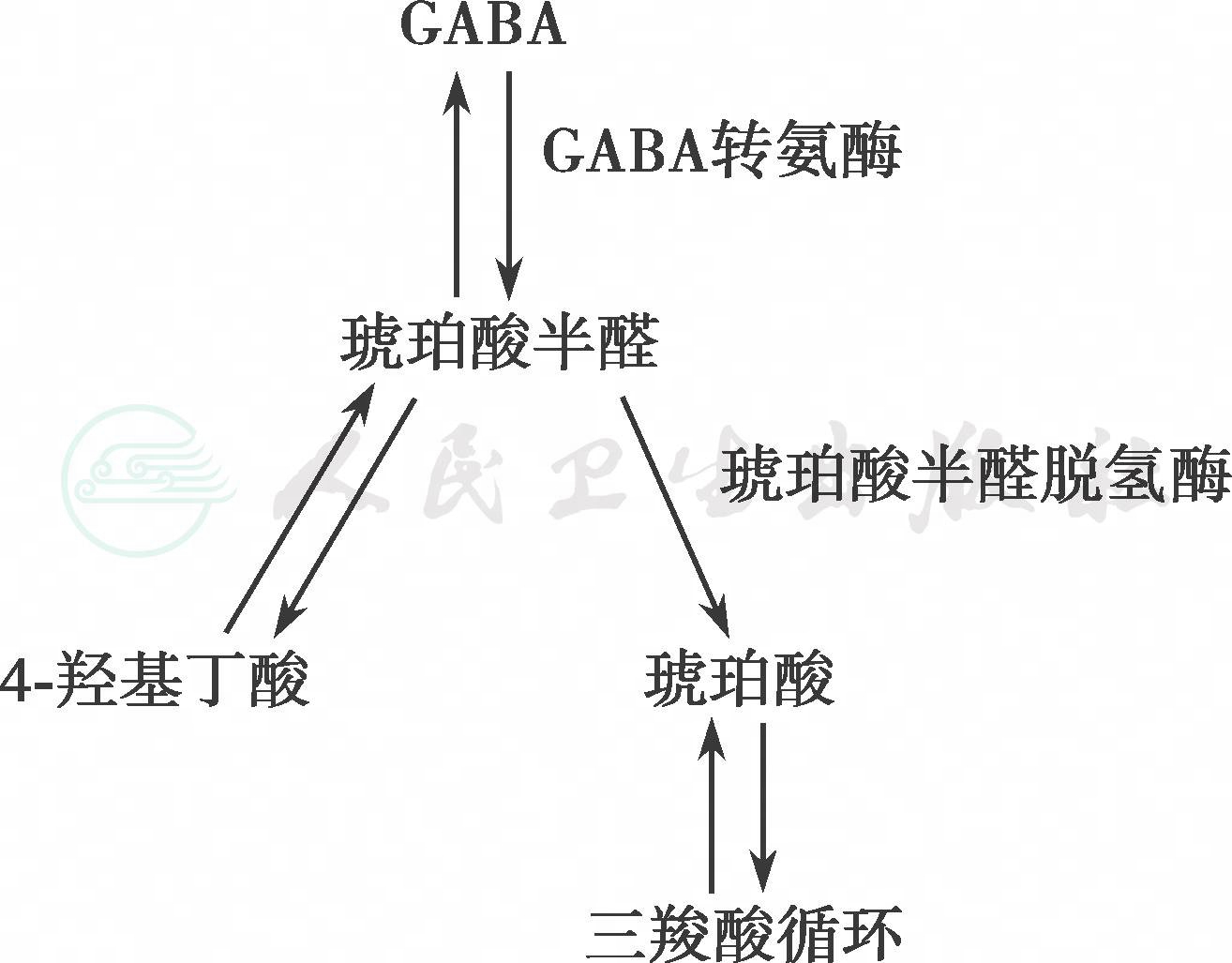
图1γ-氨基丁酸代谢途径
引自:儿童神经病学(第3版).第3版.ISBN:978-7-117-32062-7.主编:
GABA是主要的中枢神经系统抑制性递质,可调节多种神经递质的活性,包括多巴胺、血清素以及去甲肾上腺素。GABA的合成通过一步反应,由前体谷氨酸盐经过谷氨酸脱羧酶作用形成。GABA的代谢通过连续的转氨作用和氧化作用分别产生琥珀半醛和琥珀酸,琥珀半醛在SSADH催化作用下转化成琥珀酸,而在琥珀半醛还原酶催化作用下转化成4-羟基丁酸。
对SSADH缺乏症患者进行研究,发现患者都是ALD5A1基因的突变纯合子,目前共发现48个突变位点,几乎所有的错义突变都会导致SSADH活性至少减少5%,提示其他的修饰因子对疾病发病机制也有重要作用。
和正常人比较,SSADH缺乏症患者脑中4-羟基丁酸含量提高了30倍,GABA含量提高了2~4倍。GABA通过结合受体GABAB起作用,此外还有GABAA受体和GABAC受体。在此疾病中GABAB受体是最重要的受体,并且在GABA和4-羟基丁酸释放中起关键作用。如果GABA和4-羟基丁酸水平提高会影响GABAB受体功能,会出现强直阵挛发作。在细胞内通路,4-羟基丁酸通过GABAB受体抑制有丝分裂原激活蛋白(MAP)激酶活性。有丝分裂原激活蛋白激酶在许多生理变化中起必要作用,包括调控细胞分裂和细胞分化,因此在SSADH患者中出现此信号通路下调,另外4-羟基丁酸作用于有丝分裂原激活蛋白激酶会影响髓磷脂表达,髓磷脂是神经元外侧的脂质,包裹轴突,起到保护和绝缘的作用。正常的髓鞘形成对于在神经细胞间传递电信号或数据具有决定性作用。当鞘磷脂受损,会出现很多神经性疾病。SSADH缺乏症患者中谷氨酸盐代谢也受到影响。子通道型谷氨酸盐受体包括N-甲基-D天冬氨酸和α-氨基-3-羟基-5甲基异唑-4丙酸/钾盐镁矾受体,高水平的4-羟基丁酸会抑制这两个受体的作用,并且改变谷氨酸刺激的突触传递。谷氨酸盐减少和GABA升高会破坏谷氨酸盐-谷氨酸酯穿梭作用,会破坏谷氨酸盐的体内平衡,导致谷氨酸刺激作用和GABA抑制作用平衡被破坏,引起抽搐发作。琥珀半醛具有活性羟基,会增加氧化应激作用,这个应激作用会在脑组织中形成自由基,导致次生细胞损伤和死亡。同时氧化应激作用也会导致纹状体多巴胺的减少,导致病理改变。
正常生理状态下,脑内主要的抑制性神经递质GABA,在GABA转氨酶作用下降解成琥珀酸半醛,然后经SSADH催化进一步代谢形成琥珀酸进入三羧酸循环。当SSADH活性下降,琥珀酸半醛则通过琥珀酸半醛还原酶生成GHB,造成GHB在尿液、血清、脑脊液中大量蓄积。而过量的GHB对神经系统产生毒性作用,主要影响GABA、多巴胺、血清素、乙酰胆碱等多个神经递质系统。目前认为GHB和GABA均参与4-羟基丁酸尿症的病理过程,发病机制涉及胶质细胞神经末梢的谷氨酸-谷氨酰胺循环的失衡。
1.颅脑MRI检查
双侧苍白球对称性长T1、长T2信号是琥珀酸半醛脱氢酶缺乏症的特征性改变(图2),部分患者还可见到大脑与小脑蚓部萎缩,皮层下白质、齿状核、脑干病变。
2.EEG检查
可有背景慢化和痫样放电。
3.尿代谢筛查
尿中4-羟基丁酸增高是琥珀酸半醛脱氢酶缺乏症的特异性指标,为诊断本病的重要线索。
4.琥珀酸半醛脱氢酶活性检测
外周血淋巴细胞和皮肤成纤维细胞中琥珀酸半醛脱氢酶活性降低为本病的确诊依据。
5.基因检测
ALDH5A1基因突变可进一步确诊本病。
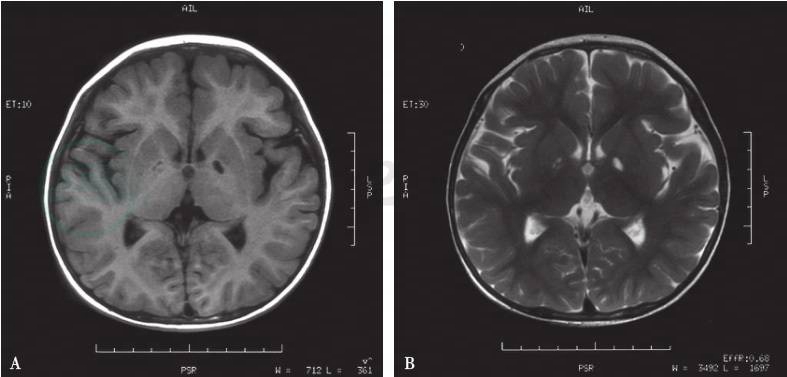
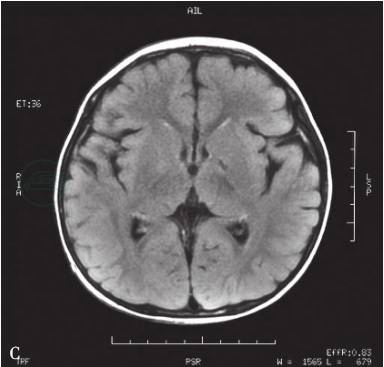
图2颅脑MRI
A.T1苍白球低信号;B.T2苍白球高信号;C.FLAIR苍白球低信号
引自:儿童神经病学(第3版).第3版.ISBN:978-7-117-32062-7.主编:
6.产前诊断
羊水中4-羟基丁酸含量检测、绒毛细胞中酶活性测定及基因突变分析已用于本病的产前诊断。
氨己烯酸为γ-氨基丁酸转氨酶抑制剂,可以减少4-羟基丁酸的产生,但有可能进一步增加γ-氨基丁酸的水平,同时其具有视野缺损的副作用,导致其在本病中的应用存在争议,有报道仅35%左右的患者应用氨己烯酸后语言、步态、癫痫发作、行为改善。癫痫的治疗可选卡马西平、拉莫三嗪等,由于丙戊酸有可能抑制残余酶的活性,不推荐使用。左旋肉碱对部分患者有效。γ-氨基丁酸受体抑制剂(SGS-742)、4-羟基丁酸受体抑制剂(NCS-382)及琥珀酸半醛脱氢酶替代治疗在鼠模型有显著疗效。另外,由于γ-氨基丁酸可以激活mTOR通路,mTOR抑制剂治疗目前也在研究中。
1.讲解遗传代谢性疾病的相关知识,减轻其精神及心理的压力。
2.遵医嘱按时、按剂量服药,注意疗效及副作用的观察。
3.遵医嘱安排病儿饮食。动物研究提示生酮饮食可以降低抽搐的发病时间和频率,减少脑电图中痫样放电,推迟共济失调发病时间,改善体重以及增加寿命,。生酮饮食是一种脂肪高比例,碳水化合物低比例,蛋白质和其他营养素合适的配方饮食。
SSADH缺陷为常染色体隐性遗传,可通过羊水细胞培养、绒毛膜绒毛活检组织、羊水检测4‐HBA,或绒毛细胞DNA基因进行测序和突变分析,进行产前筛查。









