


 收藏
收藏 已收藏
已收藏氨基酸分解代谢可产生游离氨(NH3),而氨对中枢神经系统具有毒性作用。氨通过一系列反应而转变为尿素从而达到解毒作用。由氨转变尿素需要经过Krebs-Henseleit循环。有5个酶参与尿素的合成,即氨甲酰磷酸合酶(carbamyl phosphate synthetase),鸟氨酸氨甲酰基转移酶(transcarbamylase),精氨酸琥珀酸合酶(argininosuccinate synthetase),精氨酸琥珀酸裂解酶(argininosuccinate lyase)和精氨酸酶(arginase)。第6个酶N-乙酸谷氨酸合酶(N-acetylglutamate synthetase)是CPS的活化剂,为合成N-乙酰谷氨酸所必需。上述酶缺陷导致高氨血症(hyperammonemia),患病率1/3万,为先天性遗传性高氨血症中最常见的原因。
除尿素循环过程中酶的基因突变发生缺陷外,血浆游离氨水平明显增加;还可见于其他先天性代谢缺陷。导致高氨血症的先天性代谢同缺陷病的原因有:①尿素循环中的缺陷如 CPS,N-乙酰谷氨酸合酶,OTC,ASS,ASL;②有机酸酸血症,如丙酸、甲基丙二酸、异戊酸血症;酮硫解酶缺陷、多种羧化酶缺陷、脂酰辅酶A脱氢酶缺陷(Ⅱ型戊二酸酸血症),3-羟-3-甲基戊二酸血症;③赖氨酸尿蛋白耐量减退症(lysinuric protein intolerance);④高鸟氨酸血症-高氨血症-高瓜氨酸血症综合征;⑤周期性高赖氨酸尿症伴高氨血症;⑥新生儿暂时高氨血症。
(一)一般治疗
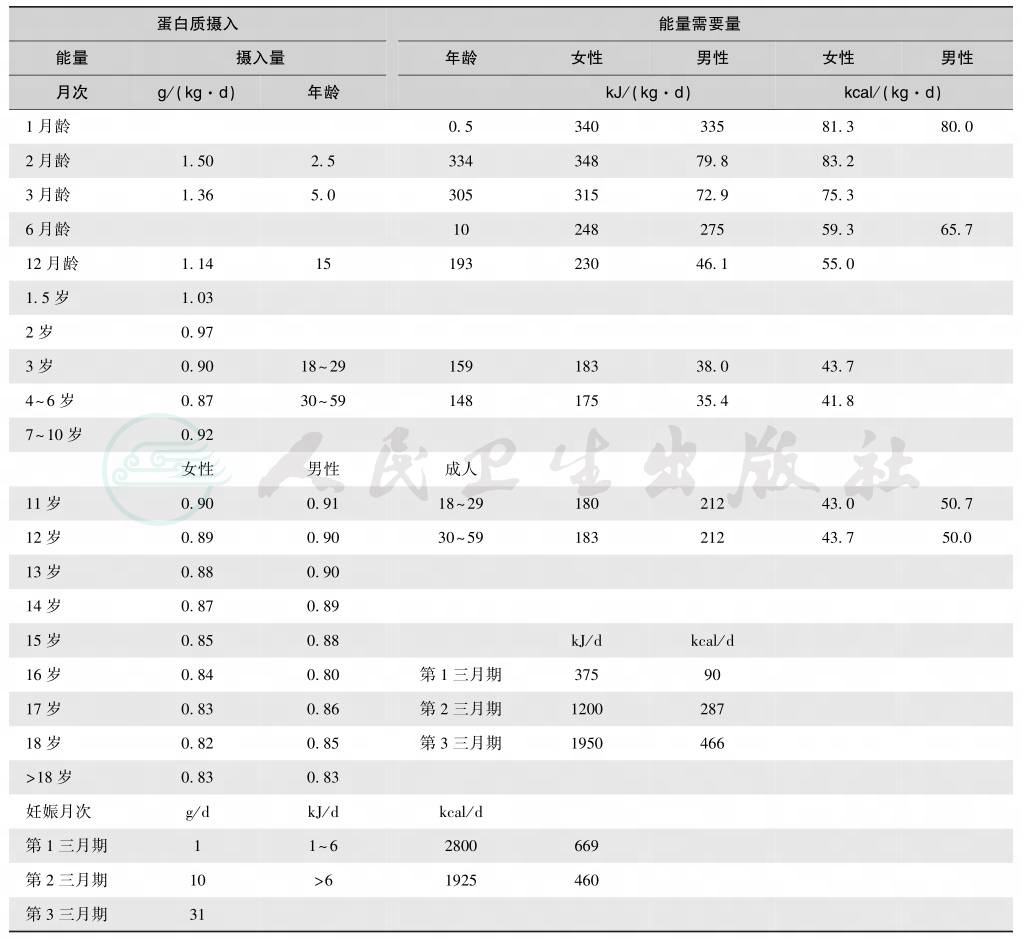
急性高氨血症及时并积极治疗,治疗目标为消除体内过多氨并提供足够热卡和必需氨基酸,以终止内源性蛋白的分解。静脉内输给足够的热卡、液体和电解质。静脉给予脂质1g/(kg·d)以保证有效提供热卡。蛋白质摄取的安全量与需要量见表3,蛋白则以最小的量0.25g/(kg·d),并以必需氨基酸为主,以防止分解代谢状态,而不增加氮负荷。有采用必需氨基酸酮酸类似物,但其有益作用未被临床应用所证实。待临床症状明显改善后宜通过鼻饲管尽早给予低蛋白营养处方0.5~1g/(kg·d)。
表3 蛋白质摄取的安全量与需要量
急性高氨血症和失代偿性尿酸循环缺陷症的药物用量见表4。婴儿急性高氨血症的治疗方案:①最初24小时,静脉提供足够热卡,液体和电解质(10%葡萄糖和静脉脂质1g/kg),加入最小量蛋白质,给予必需氨基酸混合物0.25g/kg。②起始治疗高氨血症采用下述药物:苯甲酸钠250mg/kg,苯乙酸钠250mg/kg,盐酸精氨酸 200~800mg/kg,浓度为10%溶液,加入10%葡萄糖20ml/kg溶液中在1~2小时内静脉滴注。③然后静脉持续滴注苯甲酸钠250~500mg/kg、苯乙酸钠 250~500mg/kg和精氨酸 200~800mg/kg。④若上述处理后未见血氨明显下降则可开始腹膜透析或血液透析。若在瓜氨酸血症和精氨酸琥珀酸酸性尿时,可将上述剂量加量。精氨酸不宜应用于有精氨酸酶缺陷者及有机酸酸血症所致高氨血症患儿。
(二)病因治疗
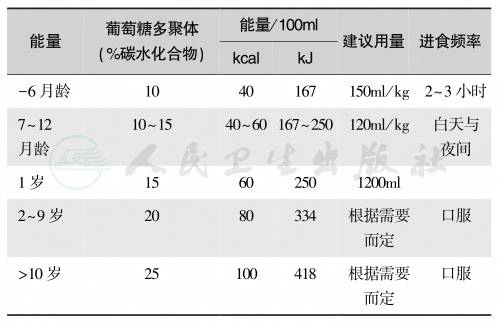
一般来说,所有患者不论哪种酶缺陷,都需要程度不等地限制蛋白摄入量1g/kg(表5)。有尿素循环中酶缺陷者应长期给苯甲酸250~500mg/kg、苯乙酸250~500mg/kg和精氨酸200~400mg/kg。OTC缺陷者,给予瓜氨酸200~500mg/kg以维持血氨水平在正常范围内。苯乙酸由于其特殊味不易为家属及患者所接受。因为苯甲酸和苯乙酸可致肉碱缺乏,补充肉碱(carnitine)是合适的,但临床效果留待证实。应尽量避免分解代谢亢进而促发高氨血症。
表4 急性高氨血症和失代偿性尿酸循环缺陷症的药物用量
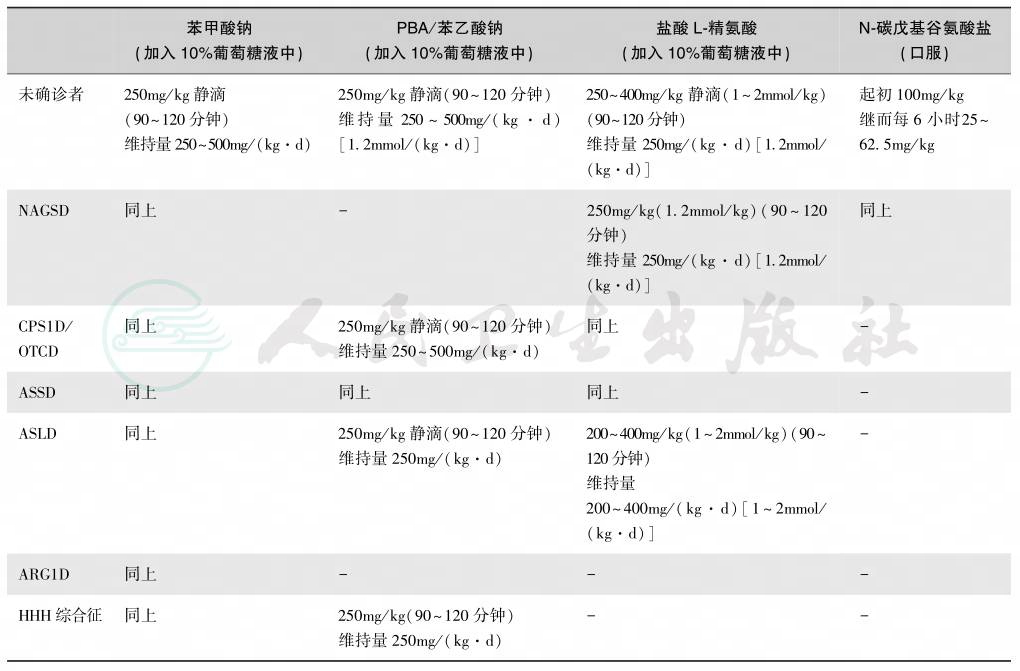
表5 儿童急症患者的无蛋白饮食治疗
1.氨甲酰磷酸合酶/N-乙酸-谷氨酸合酶缺陷症
氨甲酰磷酸合酶和N-乙酸-谷氨酸合酶缺陷症可产生同样的临床和生化改变。患病婴儿一般于出生后几日即有拒食、呕吐、嗜睡、抽搐和昏迷。CPS的晚期类型的特点为精神迟钝伴有呕吐和嗜睡发作。实验室检查示高氨血症而血浆中无特异性氨基酸增加;在继发于高氨血症患者可见谷氨酰胺、丙氨酸浓度明显增高,尿乳清酸一般偏低或缺乏。CPS缺陷的治疗类同高氨血症的治疗原则。N-乙酰谷氨酸合酶缺陷者给予氨甲酰谷氨酸可受益。故要区别两种酶缺陷,有必要做肝活检分别测定酶的活性。CPS缺陷是血常染色体隐性遗传,肝肠组织中存在此酶,由2号染色体短臂上的基因编码。N-乙酰谷氨酸合酶可由肝活检标本中测定。
2.鸟氨酸氨甲酰基转移酶缺陷症
鸟氨酸氨甲酰基转移酶缺陷症有20多个等位基因突变已被证实。杂合子女性或为轻症或无临床表现。在尿素循环异常疾病中,此种酶缺陷最为常见。起病于婴儿及儿童期,发作一般发生在高蛋白饮食、应激或感染情况下。临床表现为男性新生婴儿有严重的高氨血症。轻型见于女性杂合子及某些的男性,呈发作性表现。高氨血症发作表现为呕吐、神经异常如共济失调、意识模糊、激动和攻击性。间歇期一般良好,在发作期可有昏迷乃至死亡。智力发育可正常发展,但有轻中度智力迟钝属常见。存活者可有胆结石,机制不清。实验室检查主要为高氨血症而血中未见特异性氨基酸水平增加;谷氨酰胺及丙氨酸增加则继发于高氨血症,类同CPS缺陷。尿中有乳清酸排出过多,可区别于CPS缺陷。乳清酸在尿中沉淀成碎石。轻型在间歇期其各种异常的实验室数据可转为正常。应区别于儿童其他各种发作状态和中毒,尤其是赖氨酸尿蛋白耐量减退者其临床和生化特点类似OTC缺陷,尿中赖氨酸、鸟氨酸和精氨酸排出增加。血中瓜氨乙酸浓度增高,为赖氨酸尿蛋白耐量减退的显著特征。诊断可根据肝内此酶缺陷为特点;围生期诊断采用胎儿肝活检,近年用绒毛DNA多态性来确诊。无症状性杂合子女性携带者可采用口服蛋白质负荷而增加血氨和尿乳清酸来证实。无症状女性携带者可有轻度脑功能异常。治疗类同CPS缺陷,可用瓜氨酸取代精氨酸。在某些OTC缺陷者可采取肝移植来治疗。
3.精氨酸琥珀酸合酶缺陷症
精氨酸琥珀酸合酶缺陷症(瓜氨酸血症,citrullinemia)为常染色体隐性遗传,基因位于9号染色体长臂,来自双亲的突变基因异常的严重性并不一致,提示受累患者为复合的或双倍的杂合子。本病有明显的临床和生化异质性。临床表现范围可从轻度无症状到严重表现。新生儿型的临床表现在症状和体征方面类同CPS和OTC缺陷的严重类型,轻型可缓慢起病,有生长障碍、反复、发育延迟、毛发干脆,轻型可有发作性表现类似OTC缺陷,有些患者到20岁才出现症状。实验室类似OTC缺陷,血浆瓜氨酸浓度增高,尿中乳清酸中度增高,可有乳清酸结晶。精氨酸琥珀酸尿症患者可有血浆瓜氨酸增加,尚有精氨酸和瓜氨酸水平上升。诊断可由酶活性测定证明,可用培养的成纤维细胞检测正常的酶活性。围生期诊断依靠培养的羊膜细胞。治疗类同尿素循环中其他疾病的处理。有症状的新生儿的预后极差,轻症患者可在限制蛋白饮食后生存,即使存活者可有轻度到中等度智力发育延迟。
4.精氨酸琥珀酸裂解酶缺陷症
精氨酸琥珀酸裂解酶缺陷症(精氨酸琥珀酸尿症)患病率为1/7万活产儿。基因位于7号染色体长臂。临床和生化表现严重程度不一,新生儿型可在出生后头几日有严重高氨反应,一般死亡很高。在亚急性或晚发型主要表现为智力迟钝,伴有发作性呕吐,生长障碍和肝大;毛发异常表现为干而脆,具有特殊诊断价值。毛发异常程度较轻者亦见于瓜氨酸血症。胆结石见于某些存活者。实验室见高氨血症、肝酶中度升高,血浆谷氨酰胺和丙氨酸升高,瓜氨酸水平中度升高,血浆精氨酸琥珀酸明显增高。在氨基酸分析中,精氨酸琥珀酸位于异亮氨酸或甲硫氨酸区域,可使诊断混淆。此酶正常存在于红细胞、肝和培养成纤维细胞中,在受累胎儿的羊水中见精氨酸琥珀酸升高。治疗类似瓜氨酸血症。
5.精氨酸酶缺陷症
精氨酸酶缺陷症由常染色体隐性遗传特点,在人体有2种基因不同的精氨酸酶:一种在红细胞及肝的细胞质中表达;另一种则在肾脏线粒体中表达。细胞质的精氨酸酶在精氨酸酶缺陷者则可确定,基因定位于6号染色体长臂,其临床表现不同于其他尿素循环的一些酶缺陷,起病隐匿,婴儿在出生后头几个月或有时在几年内并无症状,以后出现进行性痉挛性剪刀样下肢麻痹。可发生舞蹈手足徐动症样运动。精神迟钝为进行性的,常见抽搐。在此疾病中一般不见严重高氨血症发作,可见肝大。实验室检查显示血浆和脑脊液中精氨酸明显增高,尿中乳清酸中度增高;血浆氨水平正常或轻度升高,尿中精氨酸、赖氨酸、胱氨酸、鸟氨酸排泄一般增加,提示胱氨酸尿诊断。尿中胍类化合物(如α-酮胍戊酸)明显增高。诊断依靠红细胞精氨酸酶测定而确定。分娩前诊断尚未能达到。治疗:无精氨酸的低蛋白饮食,给含有必需氨基酸组织的合成蛋白可使血浆精氨酸浓度明显降低,神经异常得以改善。饮食成分和每日蛋白摄取量应根据反复测定的血浆氨基酸来进行调整。苯甲酸钠250~375mg/kg可控制高氨血症。









