


 收藏
收藏 已收藏
已收藏家族性脂蛋白异常症是一组因脂质代谢酶基因缺陷所致的血脂谱异常症,主要包括家族性高胆固醇血症、家族性载脂蛋白B100缺陷症、家族性混合性血脂异常症、家族性异常β脂蛋白血症、家族性高甘油三酯血症、脂蛋白脂酶缺陷症和载脂蛋白CⅡ缺陷症。
家族性高胆固醇血症分为单基因家族性高胆固醇血症(familial monogenic hypercholesterolemia)和家族性多基因高胆固醇血症两种。杂合子异常(LDL受体突变)所致的家族性高胆固醇血症(常染色体显性遗传)的最明显表现是早发性肌腱黄色瘤。患者的血胆固醇自幼升高,并随年龄的增长而进一步升高,肌腱黄色瘤加重,同时可出现扁平黄色瘤、结节疹性黄色瘤或其他皮肤脂性瘤斑。由于纯合子异常(LDL受体突变)所致的家族性单基因高胆固醇血症亦呈常染色体显性遗传,患病个体的父母均为LDL受体突变者。因而病情重,预后不良。血胆固醇>15mmol/L(600mg/dl),有时可高达30mmol/L(1200mg/dl);多数患者早年即发生心绞痛、主动脉狭窄或冠心病,2岁即可发生心肌梗死,寿命不超过30岁。此外,杂合子LDL受体突变携带者(血胆固醇可正常)亦易发生冠心病。
(一)LDL受体-受体后信号分子突变
家族性高胆固醇血症是一种相当常见的常染色体显性遗传性疾病。本病是低密度脂蛋白受体(LDL受体,LDLR)途径(LDL-receptor pathway)变异(如 LDLR、LDLRAP1、PCSK9)所致的低密度脂蛋白代谢病,血浆总胆固醇水平和低密度脂蛋白水平升高,患者常有多个部位黄色瘤以及早发冠心病。
1.家族性高胆固醇血症
发病的原因是低密度脂蛋白受体基因的自然突变,包括缺失、插入、无义突变和错义突变。已发现数十种低密度脂蛋白受体基因突变。造成肝及外周组织细胞膜表面的低密度脂蛋白受体功能异常导致血浆总胆固醇水平和低密度脂蛋白水平升高。一般可分为五种类型:①Ⅰ类突变:突变基因不产生可测定的低密度脂蛋白受体,细胞膜上无低密度脂蛋白受体存在,是最常见的突变类型;②Ⅱ类突变:突变基因合成的低密度脂蛋白受体在细胞内成熟和运输障碍,细胞膜上低密度脂蛋白受体明显减少,也较常见;③Ⅲ类突变:突变基因合成的低密度脂蛋白受体可到细胞表面,但不能与配体结合;④Ⅳ类突变:此类突变是成熟的低密度脂蛋白受体到达细胞表面后虽能结合低密度脂蛋白,但不能出现内移;⑤Ⅴ类突变:低密度脂蛋白受体的合成、与低密度脂蛋白的结合以及其后的内移均正常,但受体不能再循环到细胞膜上。
杂合子家族性高胆固醇血症发生率约为1/500,典型杂合子家族性高胆固醇血症患者血浆胆固醇较正常升高2~3倍,常>7.8mmol/L(300mg/dl),低密度脂蛋白胆固醇>6.5mmol/L(250mg/dl),血浆甘油三酯不升高。但有些杂合子患者的血浆胆固醇可正常或稍升高。男性杂合子患者至45岁前后可有冠心病;而杂合子女性患者的发生年龄较男性晚10年左右。纯合子患者罕见,患者因体内无或几乎无功能性的低密度脂蛋白受体,血胆固醇显著升高,多数在15.6~26.0mmol/L(600~1000mg/dl),低密度脂蛋白浓度在 14.3~24.7mmol/L(550~950mg/dl)。 并在 10岁前出现冠心病,其特征性表现为降主动脉的广泛性动脉粥样硬化,并在20岁前死于心肌梗死。此外,因血浆低密度脂蛋白被巨噬细胞摄取,胆固醇沉积在动脉壁、肌腱和皮肤,患者几乎都伴有扁平状黄色瘤和角膜弓(胆固醇浸润所致)。
2.家族性混合性血脂谱异常症
病因未明。其主要临床特点是:①在汉族人群中相对常见;②肥胖、胰岛素抵抗、高尿酸血症和早发性冠心病;③血TG和/或胆固醇中度升高,HDL-胆固醇降低;④排除糖尿病、肾病综合征和甲状腺功能减退可能。
(二)诊断
如血浆胆固醇浓度超过9.1mmol/L(350mg/dl),家族性高胆固醇血症的诊断即可成立;若同时发现患者或其一级亲属中有肌腱黄色瘤,第1代亲属中有高胆固醇血症或家庭成员有儿童高胆固醇血症,更支持其诊断。杂合子患者的血浆胆固醇为6.5~9.1mmol/L(250~350mg/dl),并同时有上述表现之一者,亦可做出诊断。纯合子患者的诊断依据是父母有高胆固醇血症,患者在儿童时期的血浆胆固醇超过13.0mmol/L(500mg/dl),并出现黄色瘤。男性杂合子型年龄45岁可有冠心病,而杂合子女性患者发生的年龄较男性晚10年左右。纯合子患者因体内无或几乎无功能性的低密度脂蛋白受体,胆固醇水平很高,多在10岁前就出现冠心病的临床症状和体征,降主动脉易发生广泛的动脉粥样硬化,伴肌腱黄色瘤和眼睑扁平状黄色瘤。如不及时有效治疗多在20岁前死于心肌梗死。美国MedPed大纲的家族性高胆固醇血症诊断切点见表1。
表1家族性高胆固醇血症的诊断切点(血胆固醇/mmol/L)
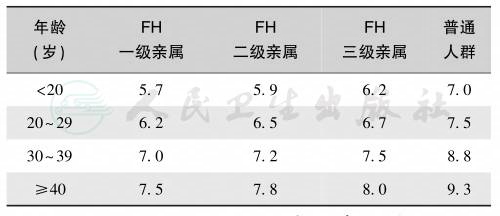
注:FH:familial hypercholesterolemia,家族性高胆固醇血症
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6.主编:
如果为单纯性高胆固醇血症,且血浆胆固醇浓度超过9.1mmol/L(350mg/dl),家族性高胆固醇血症的诊断无困难;若同时发现患者或其一级亲属中有肌腱黄色瘤、第1代亲属中有高胆固醇血症、家庭成员有儿童期就被检出有高胆固醇血症者,更支持其诊断。对于杂合子家族性高胆固醇血症,血浆胆固醇浓度为6.5~9.1mmol/L(250~350mg/dl)之间,若同时有上述表现之一者,可做出家族性高胆固醇血症的诊断,但应与家族性载脂蛋白B-100缺陷症、多基因高胆固醇血症和伴高甘油三酯血症的家族性高胆固醇血症鉴别。家族性高胆固醇血症需与家族性载脂蛋白B-100缺陷症、多基因遗传性高胆固醇血症和伴高甘油三酯血症的家族性高胆固醇血症鉴别。在儿童期,多基因遗传性高胆固醇血症者的血浆胆固醇正常,成年期后血胆固醇仅轻度升高,不伴有肌腱黄色瘤。
(三)治疗
家族性高胆固醇血症的治疗应包括低脂肪饮食、低胆固醇饮食和联合药物治疗。单纯饮食控制,血浆胆固醇降低幅度较小(5%~15%)。他汀类药物是治疗家族性高胆固醇血症患者的首选药物,如洛伐他汀、辛伐他汀等。与其他降脂药物(如胆酸螯合剂)合用可使70%的杂合子患者的低密度脂蛋白降至正常。如果本有高甘油三酯血症,可在他汀类药物的基础上,加用烟酸类降脂药物或选择性PGD2受体拮抗剂。纯合子型家族性高胆固醇血症治疗相当困难,饮食和药物治疗失败考虑定期血浆置换治疗或肝移植治疗。
家族性载脂蛋白B-100缺陷症(familial defective apolipoprotein B-100)是一种较常见的脂质代谢性疾病。据估计,人群中家族性载脂蛋白B-100缺陷症的发生率高达0.5%。载脂蛋白B-100(Apo-B100)突变造成含缺陷载脂蛋白B-100的低密度脂蛋白与受体结合障碍,影响低密度脂蛋白在体内的分解代谢,血浆低密度脂蛋白和总胆固醇升高。在正常脑组织中,细胞因子(如 TNF-α和 IL-1α/β)的表达量很低,而脂质在正常脑组织中的含量高,代谢十分活跃。卒中后,脑组织的炎性反应强烈,细胞因子对脂质代谢和其后的ROS生成起了重要作用。磷脂酰胆碱(phosphatidylcholine)和神经鞘脂(sphingomyelin)属于脂质信号物,而神经鞘脂合酶(sphingomyelin synthase)是联系糖脂和神经鞘脂代谢的关键酶。TNF-α和IL-1α/β能诱导磷脂酶A2、C、D和神经磷脂酶(sphingomyelinase)、磷脂酰胆碱合酶和神经鞘脂合酶。临床表现与家族性高胆固醇血症相似,包括血浆总胆固醇和低密度脂蛋白胆固醇浓度中度或重度升高、黄色瘤和早发冠心病。但家族性载脂蛋白B-100缺陷症所引起的血浆胆固醇水平升高的幅度低于家族性高胆固醇血症者,但较少伴有重度高胆固醇血症。部分伴肌腱黄色瘤、颈动脉粥样硬化和高血压。
根据血浆低密度脂蛋白水平增高,甘油三酯水平正常,特别是有肌腱黄色瘤和早发冠心病家族史可作出临床诊断,必要时,载脂蛋白B-100基因突变检测可予鉴别。由于家族性载脂蛋白B-100缺陷症是单基因突变所致(家族性高胆固醇血症为多个基因突变性疾病),因此,载脂蛋白B-100基因的突变检测是鉴别两者的最有效方法。
家族性混合性高脂血症(familial combined hyperlipidemia)呈常染色体显性遗传,与脂蛋白酯酶缺陷和胰岛素抵抗有关。表现为血浆胆固醇和甘油三酯水平升高及冠心病,但高密度脂蛋白往往轻度降低,伴葡萄糖耐量降低、肥胖、高尿酸血症等,但无特征性黄色瘤。病因与脂蛋白酯酶缺陷和胰岛素抵抗综合征有一定关系,其临床表现与发展规律亦与代谢综合征基本相似。氧化应激可引起动脉粥样硬化。家族性混合型高脂血症的血管氧化应激反应特别明显的原因之一是常合并有胰岛素抵抗。血脂谱异常症主要表现为血浆胆固醇和/或甘油三酯增高,高密度脂蛋白降低,常伴葡萄糖耐量降低、肥胖、高尿酸血症和早发冠心病。有中度高甘油三酯血症和/或高胆固醇血症(脂蛋白Ⅱa,Ⅱb或Ⅳ型)患者,同时存在低血浆高密度脂蛋白胆固醇血症、肥胖、胰岛素抵抗和高尿酸血症者,要高度怀疑家族性混合性高脂血症可能。但必须排除其他相关的继发性和原发性疾病(如糖尿病、肾病综合征、甲状腺功能减退等)可能。
家族性混合性高脂血症患者发生2型糖尿病的风险极高,治疗原则和方法与代谢综合征或2型糖尿病合并血脂异常基本相同。降低体重和饮食治疗有助于纠正肥胖、胰岛素抵抗和血脂谱异常症。较重患者需用药物治疗,一般可给予HMG-CoA还原酶抑制剂、烟酸、阿司匹林和胆酸,其中以HMG-CoA还原酶抑制剂较好,烟酸可加重糖耐量异常,而胆酸能升高高密度脂蛋白胆固醇,但同时使甘油三酯增高。
家族性异常 β脂蛋白血症(familial dysbetalipoproteinemia)又称为Ⅲ型高脂蛋白血症。ApoE常染色体显性突变患者罕见。多数属于ApoE常染色体隐性突变,多见于男性。家族性低β脂蛋白血症(familial hypobetalipoproteinemia)是ApoB代谢异常的常染色体显性遗传疾病,以血浆胆固醇和低密度脂蛋白胆固醇明显降低为特征。
(一)病因
大多数患者病因是由于Apo-B基因突变导致Apo-B蛋白的结构和功能异常,少数患者的病因未明。Apo-B脂蛋白降低导致血浆胆固醇和甘油三酯减少。Apo-B缺陷亦引起肠乳糜微粒形成障碍,并进一步影响脂质(包括胆固醇)和脂溶性维生素吸收,其中维生素E吸收不良导致退行性神经病变和退行性视网膜病变。
(二)临床表现与诊断
杂合子患者常见,无临床症状,偶伴有脂肪吸收障碍表现,低胆固醇血症多被意外发现,伴LDLC降低,而HDLC正常或轻度升高。发生冠心病的危险性低于正常人群。纯合子或复合性杂合子患者罕见,因脂肪吸收障碍和血浆胆固醇降低,伴吸收不良综合征、维生素E缺乏症、渐进性退行性神经病变、色素沉着性视网膜炎及棘红细胞血症。一些纯合子患者仍能产生足够的有功能的Apo-B,其病情较轻。因Apo-E基因的缺陷导致脂蛋白分解代谢的异常,其特点是血浆中聚集富含胆固醇的残体颗粒血症,高密度脂蛋白胆固醇正常,低密度脂蛋白胆固醇降低。手掌褶皱处有扁平黄瘤和在肘、膝、臀部皮肤出现黄色瘤。患者易过早发生外周血管病变和冠心病。当家族性异常β脂蛋白血症合并有Sheehan综合征时,血总胆固醇和低密度脂蛋白-胆固醇可有不同程度下降,但中密度脂蛋白-C仍明显升高。非肝病者出现掌部的结节状黄色瘤具有诊断价值。琼脂糖凝胶电泳时极低密度脂蛋白迁移到β位置与正常的β位脂蛋白重叠,形成阔β带(阔β脂蛋白症)。 血浆胆固醇[7.8~10.4mmol/L(300~400mg/dl)]、甘油三酯3.4~4.5mmol/L(300~400mg/dl)和血清胰岛素明显增高,高密度脂蛋白胆固醇正常,低密度脂蛋白胆固醇降低。手掌褶皱、肘、膝和臀部的扁平黄色瘤较常见,多数伴有早发性动脉粥样硬化、冠心病、周围血管病变、肥胖和糖尿病等。
在临床上,血浆胆固醇和甘油三酯升高者应考虑本症可能;如血浆中以富含胆固醇的β-极低密度脂蛋白和中间密度脂蛋白颗粒升高为特征。家族性低β-脂蛋白血症、无β-脂蛋白血症和乳糜微粒潴留病的鉴别见表31.3-2。极低密度脂蛋白/甘油三酯≥0.3(mg/mg)有确诊意义;结节状黄色瘤对本症有特殊诊断价值,但要首先排除肝病可能。琼脂糖凝胶电泳时极低密度脂蛋白迁移到β位置,与正常的β位脂蛋白不可分离,故形成阔β带(阔β脂蛋白症)。等电点聚焦电泳常可发现异常的Apo-E。
表2原发性低β-脂蛋白血症的遗传分型
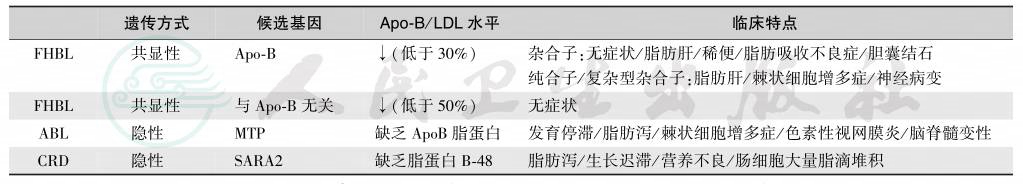
注:FHBL:familial hypobetalipoproteinemia,家族性低 β-脂蛋白血症;ABL:abetalipoproteinemia,无 β-脂蛋白血症;CRD:chylomicron retention disease,乳糜微粒潴留病
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6.主编:
血浆总胆固醇及LDLC降低往往提示本病的诊断。血浆胆固醇和甘油三酯水平极低并伴有脂肪吸收障碍时要考虑纯合子型家族性低β脂蛋白血症可能,但应与β-脂蛋白缺陷症和Anderson病(乳糜微粒滞留综合征)鉴别。Apo-B凝胶电泳或基因突变分析可确定其分子病因。杂合子型患者无症状者无须特殊处理,补充脂溶性维生素有一定意义。纯合子型患者应每天口服大剂量维生素E100~300mg/kg,以升高组织维生素E浓度,防止神经病变的发生。提高饮食中的脂肪含量(常占总热量的15%~20%)。禁忌补充中链甘油三酯(肝中毒)。血清残余脂蛋白-C(RLP-C)和甘油三酯(TG)比值(RLP-C/TG)以及Apo-E/Apo-CⅢ比值升高可用于Ⅲ型高脂蛋白血症的筛选。
(三)治疗
主要是控制体重,限制脂肪、饱和脂肪酸和胆固醇的摄入量。药物治疗主要是HMG-CoA还原酶抑制剂、烟酸和纤维素衍生物。绝经后女性Ⅲ型高脂蛋白血症可加用tibolone,因其可明显降低血TG、TC、VLDL-C和VLDL-甘油三酯水平。
家族性高甘油三酯血症(familial hypertriglyceridemia)常见,常染色体显性遗传。由于患者存在肝脏合成甘油三酯过多的代谢缺陷,使富含甘油三酯的大分子极低密度脂蛋白增多。患者多到成年才表现为持续性高甘油三酯血症,20岁以下者常无明显异常。家族性高甘油三酯血症患者常合并肥胖、高血压、高尿酸血症、糖耐量异常、高胰岛素血症和动脉粥样硬化,易发生胰腺炎与疹状黄色瘤。血浆甘油三酯升高,低密度脂蛋白和高密度脂蛋白水平降低。
鱼眼病患者多到成年才表现为持续性高甘油三酯血症,常合并肥胖、高血压、动脉粥样硬化、高尿酸血症、糖耐量异常和高胰岛素血症。患者易发生胰腺炎,四肢及躯干出现疹状黄色瘤。血浆甘油三酯多在3.4~9.0mmol/L(300~800mg/dl),当>11.3mmol/L(1000mg/dl)时,可伴有脾大,但血浆低密度脂蛋白和高密度脂蛋白降低。合并糖尿病时,常引起血浆极低密度脂蛋白明显增加,同时出现空腹乳糜微粒血症。首先应积极限制饮食中的饱和脂肪酸摄入量。降脂治疗的药物可首选贝特类药物,同时防治急性胰腺炎。合并糖尿病、甲状腺功能减退、肥胖者应特别注意原发疾病的治疗。
脂蛋白酯酶缺陷症(lipoprotein lipase deficiency)少见,常染色体隐性遗传。
(一)病因
酯酶家族包括脂蛋白酯酶、肝酯酶和内皮细胞酯酶。在内皮细胞酯酶的作用下,内皮细胞不断分泌甘油三酯和磷脂的降解物。脂蛋白酯酶(lipoprotein lipase)缺陷症是一种少见的常染色体隐性遗传性疾病。脂蛋白酯酶活性缺乏影响甘油三酯的分解代谢,导致乳糜微粒代谢受阻,乳糜微粒在体内积聚。酯酶成熟因子-1(lipase maturation factor-1,LMF-1)是细胞内质网的一种膜结合蛋白,其主要功能是使脂蛋白酯酶(亦可能包括其他脂蛋白,如肝酯酶和血管内皮酯酶等)形成合适的折叠与空间结构(成熟)。LMF-1突变后,不能形成酯酶二聚体,引起混合型酯酶缺陷症(combined lipase deficiency)和高甘油三酯血症。脂蛋白酯酶位于肝外组织的毛细血管腔的内皮表面。正常情况下,乳糜微粒和内源性极低密度脂蛋白中的甘油三酯水解依赖于脂蛋白酯酶及其激活剂载脂蛋白CⅡ。因此,此代谢途径中的任何异常都将导致严重的高甘油三酯血症和乳糜微粒血症。由于脂蛋白酯酶活性降低导致乳糜微粒代谢受阻,乳糜微粒在体内的积聚。患者在婴儿或儿童期不能耐受含脂肪食品,反复发作性腹痛(胰腺炎),且伴有周期性疹状黄色瘤。可出现肝脾大和视网膜脂血症。血浆外现呈奶油样状,乳糜微粒甘油三酯明显升高。婴幼儿反复发作腹痛伴血浆呈乳状时,应高度怀疑此症。
(二)临床表现与诊断
患者在婴儿或儿童期常出现脂肪食物不耐受表现,常在进食脂肪或饱餐后发生反复发作性腹痛(胰腺炎),较重患者有典型急性胰腺炎发作史。慢性乳糜微粒血症导致的疹状黄色瘤主要集中在臂、肘及膝部伸侧,可呈周期性发作。黄色瘤直径1~2mm,高出皮肤,病理活检可见含有大量甘油三酯和胆固醇酯的成串巨噬细胞。患者可出现肝脾大,其内含有大量含脂肪的巨噬细胞。眼底检查可见视网膜苍白(视网膜脂血症)。婴幼儿反复发作腹痛伴乳状血浆时,应高度怀疑此症。年轻人禁食12小时后出现脂血血浆也提示脂蛋白酯酶缺陷症可能。静脉输入肝素后血浆脂蛋白酯酶不增加可以确诊。血浆乳糜微粒明显升高,甘油三酯常在11.3mmol/L(1000mg/dl)以上,血浆外现呈奶油样,置于4℃冷藏12小时,血浆表层漂浮一层白色物。
(三)治疗
采取饮食治疗,严格限制饮食中脂肪摄入量(占总热卡10%左右)。当患者接受严格的低脂肪或无脂肪饮食时,疾病的症状和体征可消退。必要时,可选择贝特类或他汀类药物治疗。抗脂肪生成的基因治疗有望成为脂蛋白酯酶缺陷症治疗的新途径。
载脂蛋白CⅡ(apolipoprotein CⅡ)缺陷症是一种少见的常染色体隐性疾病,发病率低于1/100万。Apo-CⅡ是脂蛋白酯酶的激活因子,Apo-CⅡ缺陷导致功能性脂蛋白酯酶缺陷,脂蛋白酯酶不被激活,甘油三酯分解障碍,血中乳糜微粒蓄积。纯合子患者由于载脂蛋白CⅡ遗传性缺陷,血清甘油三酯显著升高,血乳糜微粒、极低密度脂蛋白聚集(Ⅰ型和Ⅳ型高脂蛋白血症)或两者同时存在(Ⅴ型高脂蛋白血症),类似脂蛋白酯酶缺陷症的乳糜微粒血症。本症的临床表现与脂蛋白酯酶缺陷症相似,纯合子患者发病年龄一般较家族性脂蛋白酯酶缺陷症晚,患者多在婴幼儿期发病,表现为反复发作性腹痛、胰腺炎、网膜色素变性和发育障碍;疹状黄色瘤和肝脾大不常见,但可伴嗜多染红细胞性贫血。杂合子患者的甘油三酯可正常或轻度增高,一般不发展为胰腺炎。
确诊有赖于极低密度脂蛋白载脂蛋白凝胶电泳(显示缺陷载脂蛋白CⅡ),注入肝素后血浆脂蛋白脂酶活性仍缺乏,而加入正常血清(含载脂蛋白CⅡ)后,脂蛋白酯酶恢复正常可诊断为本症。本症的治疗与脂蛋白酯酶缺陷症相似,严重胰腺炎患者可输注正常人血浆。









