


 收藏
收藏 已收藏
已收藏(一)急性应激
1.外源性应激因素
外源性应激因素很多,常见的因素有:①过冷和过热;②烧伤;③放射性损伤;④长期剧烈运动;⑤毒物中毒、药物中毒或病原微生物。
2.特殊应激因素
主要有高热、剧烈胸痛、腹痛、缺氧、呼吸困难、窒息、呕吐、腹泻、急性出血、昏迷、抽搐、呼吸窘迫综合征、急性代谢紊乱综合征(酸中毒、碱中毒、高钠血症、低钠血症、高钾血症、低钾血症、高钙血症和低钙血症等)、急性创伤、烧伤、手术、麻醉、休克和脑血管意外等。长期以来,人们偏重于上述应激因素对机体的各种损害的研究,随着医疗技术的迅速发展,抢救的成功率也不断提高;但人们较少注意应激本身对机体的致病作用和机体反应异常所导致的各种不良后果。
(二)慢性应激
所谓慢性应激系指机体受到一种或多种长期的不良刺激的一种病理现象。应激原多种多样,可为感染、炎症、创伤、出血、中毒、器官功能衰竭或精神刺激等。在慢性不良刺激或病理因素作用下,机体处于相对高代谢状态,并调动免疫系统和内分泌系统做出相应反应。慢性应激是相对于急性应激而言的,但病因有所不同,例如噪声和制动主要引起慢性应激,慢性感染和代谢紊乱也主要以慢性应激反应为主。慢性应激的特点是神经系统反应不明显,而且由于应激性质和部位的差异,免疫系统和内分泌系统的反应不同,其特点和程度也不相同。一般来说,机体的内分泌系统反应规律是糖皮质激素分泌增多,T3下降,rT3升高,性腺类固醇激素分泌减少,糖分解增多。免疫系统的反应以感染性和自身免疫性或免疫缺陷性疾病较突出,表现为抗体水平升高或自身抗体增多,免疫细胞浸润和组织免疫性炎症反应等。细胞因子的作用增强,常形成免疫炎症-细胞因子间的恶性循环,可导致器官功能障碍或衰竭。
慢性应激引起的各种躯体和神经精神病变统称为应激相关性疾病(stress-related disease)[3,4],这些疾病常常不被人们重视。例如,咀嚼(mastication,chewing)是慢性应激的一种伴随行为习惯,而且咀嚼可激活下丘脑-垂体-肾上腺轴与自主神经功能,应激性咀嚼可降低应激引起的高皮质酮血症、高皮质醇血症和高儿茶酚胺血症,促进应激相关因子(如神经营养因子与NO)表达,减轻海马和下丘脑的应激性损伤。
亚急性应激的概念和定义较含糊,在临床上亦很难与慢性应激或急性应激严格区分。亚急性应激可能是急性应激向慢性应激过程过渡的一个病理阶段,其与慢性应激的区别要点是神经系统的调节作用在亚急性应激反应中仍较突出,表现为交感神经兴奋,儿茶酚胺和兴奋性氨基酸等对机体的损伤作用较明显。急性和慢性应激与临床上的急性疾病及慢性疾病是基本一致的,各种原因所致的急性或慢性应激的内分泌变化规律也基本相同。慢性应激常被人们忽视,一些貌似健康者,可因体内存在某种慢性疾病和慢性功能紊乱而存在长期的慢性应激反应。慢性应激的病理特点是氧化应激[4],见于许多慢性疾病中,如蛋白-营养不良症、老年性痴呆、帕金森病(Parkinson disease)及其他慢性消耗性与慢性感染性疾病等。氧化应激的病因与谷胱甘肽的代谢失常有关。
(三)创伤与应激障碍
创伤后应激障碍(posttraumatic stress disorder,PTSD)是在创伤性事件后出现的一种临床综合征,多见于儿童和青少年女性。包括心理障碍(如担心死亡、严重伤害、自身或他人的身体威胁),并导致强烈的无助与恐惧,表现为过度警觉、事件重温以及回避与事件相关的不良刺激等。长时间暴露于创伤性事件,增大了创伤后反应的可能性;患儿行为紊乱、精神不集中、睡眠障碍、噩梦、情绪沮丧和焦虑不安。重大手术后出现PTSD与以下因素相关:①ICU住院时间;②儿童住院期间,其周围患者的病情;③住院期间与父母分离;④接受侵入性操作;⑤创伤后遗症。许多生活事件可触发生理情绪反应,回避触发意识自身疾病的相关因素,使治疗依从性降低。据报道,10%~17%的心脏移植患者可出现创伤后应激综合征。术后第1年的发病率特别高,移植手术后前3年的累计发病率达17%。术后第1年内发生PTSD是长期并发症与病死率的预测因素。患者常通过闪回(flashbacks)或噩梦,再次经历创伤性事件;为了避免联想所触发的经历,患者错过预约门诊,漏服免疫抑制剂。另一方面,活性氧是导致某些神经精神疾病(neuropsychiatric disorder)如双相性精神病(bipolar disorder)的病理生理基础[5],而良好的精神心理环境是预防精神心理应激和其他应激相关性疾病(stress-related disorder,SRD)的必要条件,而以精神创伤为重点的认知-行为疗法是当前最为有效的方法,反复可采用药物联合心理与认知疗法[5,6]。
(四)细胞应激
细胞氧化应激的病因很多,其中主要有异体移植排斥反应、缺血、缺氧、炎症、中毒、电解质平衡紊乱、细胞凋亡及剧烈运动等。酒精中毒和铁中毒等也是常见的细胞氧化应激的应激原。除上述病因外,在医用植入物表面、生物膜表面和血液透析膜表面,甚至在血浆分离过程中均可产生过量的自由基。例如在血液透析过程中,红细胞的自由基清除酶被激活。由于成熟红细胞无细胞核,此酶的活性不能在基因水平进行调节,而粒细胞可通过氧化还原反应,激活氧化应激反应,故粒细胞的FRSE可更好地反映机体的氧化应激状态。
败血症休克、全身性炎症反应综合征(systemic inflammatory response syndrome,SIRS)和急性呼吸窘迫综合征的病死率很高,这些疾病的发展过程有许多相似之处。主要病理生理变化是血管张力调节紊乱(如低血压、外周血管扩张、对补液和血管收缩药有抵抗以及肺高压等),其最终后果是组织细胞的凋亡[7,8]。现认为,其病变过程亦主要与急性氧化应激有关,包括缺血再灌注损伤、氧化应激、衰老和急慢性中毒等。例如,在炎症性肠病时,出现细胞的内质网应激,这种应激反应的一个重要特点是出现非折叠型蛋白反应(unfolded protein response,UPR),即蛋白质在执行功能之前不能自行折叠成有效的空间结构,因而很容易被自噬。肠道的Paneth细胞和杯状细胞(goblet cell)因为高度依赖于蛋白的折叠来维持细胞的功能,所以特别容易发生内质网应激,并由后者引起肠道炎症[9]。
增龄性肌肉消耗和肌肉减少的发病机制本质事实上是细胞炎症反应和氧化应激的后果,而运动和营养治疗可能逆转氧化应激而减缓肌肉的消耗速度[10]。同样,高血压对血管内皮细胞也是一种氧化应激[7-10]。
各种急性应激对机体的影响不同,但机体对急性应激的反应则有许多共同之处:①交感神经兴奋,机体的应激能力增强,心、肺和脑等器官的活动加强,耗氧量增加,心率加快,心排血量增多;②交感神经-肾上腺髓质活动增强,儿茶酚胺释放增多;③下丘脑释放CRH、GHRH、AVP和TRH等增多,GH、肾上腺皮质激素和甲状腺素分泌增加;④血糖因拮抗胰岛素作用的激素分泌增多及糖原分解增强而升高;⑤失水时,AVP、肾素、血管紧张素和醛固酮的分泌亦增加,是导致水钠潴留的主要原因,有时也导致高肾素性低醛固酮血症。
(一)急性应激与神经-内分泌功能
应激系统的神经内分泌系统由下丘脑-垂体-肾上腺(HPA)、延髓交感神经、肾上腺髓质和副交感神经组成。主要包括下丘脑和脑干的CRH细胞、AVP细胞、旁神经原细胞(paragiganto-cell)、脑髓质腮旁核(parabranchial nuclei)细胞、蓝斑(locus ceruleus,LC)细胞和其他去甲肾上腺素能细胞等。下丘脑为肾上腺素能神经末梢的密集区,主要接受来自延髓腹外侧A1区和背中线A2区及部分来自蓝斑的去甲肾上腺素能神经支配。因此下丘脑的CRH细胞在去甲肾上腺素的刺激下持续性兴奋,释放过多CRH。另一方面,兴奋性氨基酸为HPA轴的重要中枢调节因子,兴奋CRH细胞的作用途径可能与N-甲基-D-门冬氨酸(N-methyl-D-aspartate,NMDA)受体的介导有关。
1.HPA轴变化
在应激过程中,下丘脑以外的边缘系统、前脑基底部、脑干交感系统和脊髓等可分泌CRH,其在协调应激反应中的作用广泛。在应激系统中,中枢的蓝斑/去甲肾上腺素神经元(LC/NE)和CRH神经元两者之间存在着反射性相互作用。室旁核的CRH神经元和脑干的NE神经元均有自身超短负反馈的神经纤维联系和功能联系,分别通过突触前CRH及α2-去甲肾上腺素能受体抑制CRH及儿茶酚胺的分泌。γ-氨基丁酸/苯二氮䓬(γ-amino-butyric-acid/benzodiazepine,GABA/BZD)及阿片肽样神经元系统(opioidpeptide-neuronal-system)的抑制性传入信号产生应激行为和适应性反应。另外,P物质和NPY亦有相互作用。P物质抑制CRH神经元而兴奋LC/NE交感系统。应激时,CRH和AVP呈比例增加,慢性炎症和疼痛时中枢神经的P物质升高,使机体状态在应激系统的激活中得到适当的改善。
各种急性应激刺激传入中枢神经系统,被神经元整合后,兴奋CRH释放的神经递质增多,CRH分泌增加,HPA轴被兴奋,肾上腺糖皮质激素释放入血,血皮质醇升高。同时还可见肾上腺血流增多、细胞肥大和增生,线粒体增加,脂质体减少。但术式不同和麻醉方式不同,反应也不尽相同。一般血皮质醇在术前即开始升高,术后2小时达高峰值(增加3.8倍),术后2天后逐渐恢复正常。在ICU病房,有70%以上的败血症和心源性休克患者存在“相对性肾上腺皮质功能不足”,这是主张应用小剂量糖皮质激素的原因和依据。但是,近年对这一理念有了较大质疑。部分患者的相对性肾上腺皮质功能不足是一种保护性反应,没有使用大剂量糖皮质激素的必要,或者说,甚至是有害的[10,11]。交感神经-肾上腺髓质-肾上腺皮质轴主要参与亚急性和慢性应激的肾上腺皮质功能调节。交感神经兴奋时,肾上腺髓质可释放肾上腺素、去甲肾上腺素、多巴胺、血清素、乙酰胆碱、脑啡肽、CGRP、VIP、CRH、PACAP、ANP 和 AM 等,并在免疫细胞的协助下,促进肾上腺皮质合成和释放糖皮质激素、生长因子和细胞因子。肾上腺糖皮质激素的调节可分为CRH/ACTH依赖性和非CRH/ACTH依赖性两条途径,自主神经系统对应激反应能迅速广泛地控制心血管、呼吸、消化道、肾、内分泌和其他系统,这些器官均由交感神经系统、副交感神经系统或两者共同调控。副交感系统有拮抗和消除交感神经的作用,在应激反应中副交感神经的兴奋性增强以拮抗交感神经的功能。来自脊髓的神经元的节前纤维将应激刺激传入交感神经支配的周围器官。交感神经节两侧链的神经突触与交感神经元的节后纤维共同支配血管平滑肌、心脏、肾、肠道、脂肪和其他许多器官。交感神经系统通过肾上腺髓质也将肾上腺素和去甲肾上腺素等分泌到体液和血循环中。自主神经除分泌乙酰胆碱及去甲肾上腺素两种神经递质外,还释放多种神经肽、ATP、一氧化氮和炎症脂质。CRH、NPY、生长抑素和甘丙肽位于去甲肾上腺素能神经元内,VTP、P物质和降钙素基因相关肽(CGRP)共同存在于胆碱能神经元内。交感神经节的节前纤维的神经递质和神经元内短的纤维传导也介导神经肽的释放。
非CRH/ACTH依赖性肾上腺糖皮质激素调节的生理和临床意义可归纳为:①肾上腺皮质的非CRH/ACTH调节途径是生理和病理情况下(包括应激)均发挥作用的内分泌调节机制。正常情况下,维持基础糖皮质激素分泌和肾上腺皮质与髓质功能。②由于非CRH/ACTH依赖性糖皮质激素分泌,患者往往出现血ACTH/皮质醇分离现象,即ACTH正常甚或下降,血皮质醇升高。临床上遇见这种分离现象时,要想到患者存在慢性或亚急性应激(如自身免疫性疾病和慢性感染等)的可能。③给患者做地塞米松抑制试验,血皮质醇无抑制反应。同理,患者对甲吡酮亦无兴奋反应。④新生儿、老年人、糖尿病及其他全身性疾病患者,尚可有血雄激素/皮质醇的分离现象,表现为血雄激素降低,血糖皮质激素升高,其发生机制未明,亦可能与非CRH/ACTH依赖性糖皮质激素的释放有关。⑤临床上鉴别CRH/ACTH依赖性和非CRH/ACTH依赖性皮质醇升高较困难,如ACTH正常或下降、皮质醇升高和胰岛素低血糖-地塞米松抑制联合试验(低血糖能增加而地塞米松不能抑制皮质醇的分泌)有助于非CRH/ACTH依赖性皮质醇分泌增多的诊断。选择性1型CRH受体和3型AVP受体阻滞试验亦有助于鉴别,这些阻滞剂可抑制ACTH介导的皮质醇分泌而对非CRH/ACTH依赖性皮质醇分泌无抑制作用。
2.交感-血管紧张素系统变化
AT-2既是肾素的作用底物又是神经递质,大脑皮质、下丘脑、交感神经元和AT-2神经元在脑内分布广泛。当应激使交感神经兴奋时,中枢神经的AT-2释放明显增多,并进一步促进儿茶酚胺、AVP和CRH等分泌,故AT-2在脑内起加强、扩增和易化中枢交感神经兴奋作用来提高机体的急性应激能力。在交感神经末梢,释放的去甲肾上腺素通过β受体使肾素-血管紧张素系统活动增强。而且,AT-2又可通过交感神经末梢的突触前受体,使去甲肾上腺素进一步释放,形成AT-2和去甲肾上腺素间的恶性循环(正反馈)。在肾上腺,AT-2促进醛固酮分泌及髓质激素释放,进一步促进各种应激激素分泌。醛固酮的作用分为基因依赖性和非基因依赖性两个方面。非基因依赖性作用可能是靶细胞膜结合物(可能为膜受体)作用,调节Na+反极转运体(antiporter)和胞内第二信使系统,改变血管平滑肌细胞和内皮细胞等的活动而出现血压升高和钠潴留等应激反应,而且还参与了急性应激时细胞内氧化调节过程。在慢性应激情况下,常因应激反应引起肾素过度分泌,但由于交感神经兴奋也抑制肾上腺皮质醛固酮合成的后期步骤而阻滞了醛固酮的合成和分泌,导致高肾素性低醛固酮综合征。
3.下丘脑-腺垂体-甲状腺(HPT)轴变化
急性应激时,由于交感神经兴奋,下丘脑TRH分泌增多,通过TSH刺激甲状腺,释放较多甲状腺激素入血,代谢率加速,以适应机体代谢的需要。如果应激时间延长,或因为某些原因病情危重,HPT轴功能往往处于抑制状态。如急性心肌梗死、高血压危象、脑血管意外、急性心力衰竭、急性肺功能衰竭、急性颅内高压和中毒性休克等患者,血清甲状腺激素变化有一定规律性。通常随着病情进展,血T3和T4下降,rT3逐渐升高,T3的下降与rT3增高呈反变关系;至疾病恢复期,T3、T4和rT3逐渐恢复正常。故有人认为,血T3和rT3是判断病变严重程度和预后的良好指标。相对血液中的T3和T4浓度变化来说,脑组织中的T3变化最为明显(T3显著增加),这是因为脑组织Ⅱ型5'-脱碘酶对急性应激十分敏感所致。
4.下丘脑-腺垂体-性腺轴变化
应激时,性腺轴被HPA轴抑制。HPA轴直接或通过弓状核POMC神经元刺激β-内啡肽和CRH,在弓状核抑制促性腺激素释放激素神经元。另一方面,糖皮质激素抑制GnRH和LH的分泌,使性腺类固醇激素的靶组织产生抵抗。在炎性应激时,炎性细胞因子也可抑制生殖功能,而瘦素起允许和激活作用。慢性HPA轴的激活引起性腺功能的抑制(如赛跑运动员和芭蕾舞蹈演员),其夜间血皮质醇和ACTH及尿游离皮质醇增高,ACTH对外源性CRH的反应迟钝。男性LH、睾酮和雌二醇水平均低,女性无月经。HPA轴的活性增高引起的病理生理变化有:①抑郁、神经性厌食和营养不良;②烦躁-强迫症;③烟(酒)戒断综合征;④糖尿病或代谢综合征;⑤精神性矮身材;⑥库欣综合征(Cushing syndrome);⑦胃肠疾病、纤维肌肉疼痛和病态甲状腺综合征等。尿中皮质醇排出量增高,对外源性CRH反应减低,脑脊液CRH升高。
无论在急性、亚急性或慢性应激情况下,HPA轴兴奋均可导致HPG轴的功能抑制。在临床上,引起神经精神症状、月经紊乱、闭经、性欲减退、阳痿、精子生成减少和不育症等,其中以生育期女性的HPG轴功能紊乱表现最为突出。应激所致性腺功能减退或功能紊乱的原因很多,以下丘脑GnRH的脉冲性分泌功能丧失最明显。应激状态下,HPG轴功能抑制主要与HPA轴兴奋有关,表现在:①应激性闭经、神经性厌食或剧烈运动妇女的皮质醇分泌增多,昼夜节律仍存在,脑脊液中CRH浓度升高;②使用外源性CRH后,卵巢切除动物表现为GnRH和LH的脉冲性分泌消失;③使用IL-1和内毒素等应激物质后,HPA轴被兴奋,同时可见HPG轴功能抑制(LH和FSH的脉冲性节律消失,分泌量下降);④位于GnRH脉冲发生器附近的β-内啡肽分泌增多,使用阿片样肽受体拮抗剂则使GnRH脉冲性分泌功能迅速恢复,而纳洛酮或纳曲酮可拮抗应激对HPG轴的抑制。这提示,β-内啡肽至少是联系HPA轴(CRH)与HPG轴(GnRH)的应激性调节物(神经调质)之一。
禁食时间过长也是一种病理性刺激。禁食时,胃分泌一种神经调节物质,通过迷走传入纤维到达下丘脑的室旁核(PVN),兴奋去甲肾上腺素能神经,CRH分泌增加再抑制GnRH的释放,雌激素则兴奋这一系统。下丘脑的PVN和A2区为雌激素的负反馈作用部位,禁食时该部位的雌激素受体数目增多,故禁食时雌激素可抑制LH的分泌。因此,可认为禁食首先引起去甲肾上腺素的分泌,促使下丘脑雌激素靶细胞中的受体数目增多,由胃部而来的传入神经冲动对去甲肾上腺素的敏感性增加。在雌性动物中,应激性刺激对性腺功能的作用还受卵巢类固醇激素的调节,但其对PVN和A2区的反馈作用与通常的“负反馈作用”机制不同,因为这种反馈信息不是来源于卵巢而是来源于环境因素,如应激。由于GnRH分泌下降可致垂体LH释放明显减少,如持续时间过长,可导致闭经或性欲减退。男性血睾酮下降,但对性腺活动的影响不明显。应激时神经系统、HPA轴和HPG轴的主要变化见图1。
(二)中枢神经对急性应激的反应
急性出血者有失血性休克、微循环障碍和神志改变等表现;重症创伤者有剧烈疼痛、功能障碍和器官组织损伤等表现;急性缺氧(低张性)时,动脉血氧分压、氧含量和血红蛋白氧饱和度降低等。由于病因不同,其临床表现各异。本节主要讨论神经-内分泌方面的临床特征。具体表现为:①神经系统表现,主要表现为交感神经兴奋,如心率加快、心搏增强、内脏血管和皮肤血管收缩,支气管平滑肌舒张、肺通气量增大,胃肠活动和消化液分泌功能减弱,瞳孔扩大和眼肌收缩,汗液分泌增多,糖原分解,儿茶酚胺分泌增多等;②内分泌系统表现,应激激素和中枢神经递质释放、HPA轴、交感神经-肾上腺髓质、甲状腺轴与交感-血管紧张素系统兴奋,而性腺轴被抑制。
1.应激激素和中枢神经递质分泌
应激激素(stress hormone)一般系指应激状态下激素分泌细胞兴奋、合成和大量分泌入血的一类激素(图1)。应激时,血浆浓度明显升高的激素主要有:①下丘脑-垂体激素,如CRH、AVP、β-内啡肽、神经肽Y、ACTH、β-溶脂素、PRL、GH和褪黑素等;②肾上腺皮质和髓质激素,如皮质醇、醛固酮、肾上腺素、去甲肾上腺素、多巴胺和肾上腺髓质素(AM);③肾素和血管紧张素;④旁分泌激素和细胞因子,如前列腺素、激肽、IL-1、IL-6、TNF、兴奋性氨基酸、自由基和应激活化蛋白激酶(stress-activated protein kinase,SAPK)等。旁分泌应激激素的释放只在局部组织起作用,血中激素浓度不一定升高。这一类应激激素的主要释放部位在中枢神经系统。例如,应激促进下丘脑及其他脑区去甲肾上腺素和兴奋性氨基酸的释放,并进一步兴奋 HPA 轴功能[11,12]。
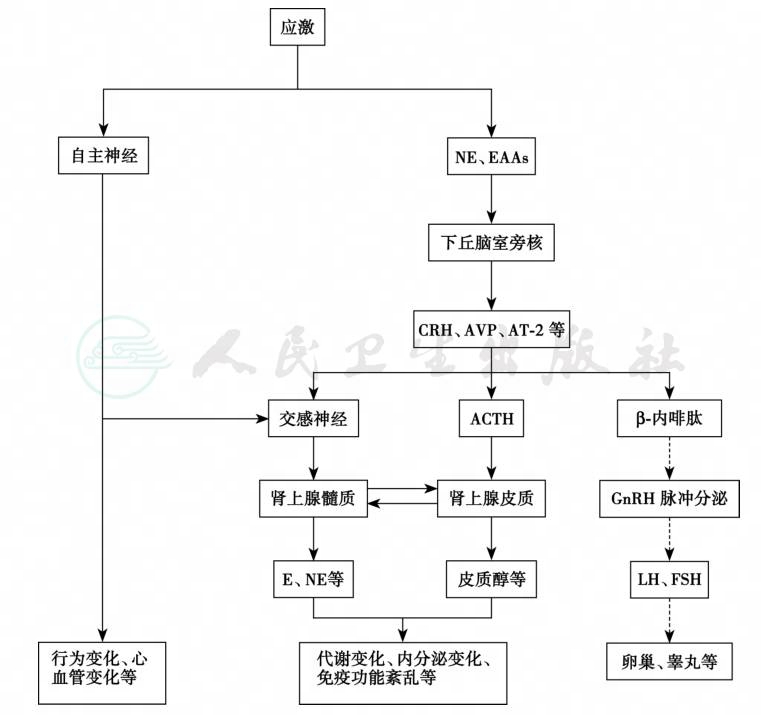
图1应激对神经、内分泌和免疫调节功能的影响
实线箭头表示兴奋,虚线箭头表示抑制;E:肾上腺素;NE:去甲肾上腺素;EAA:兴奋性氨基酸类物质;AVP:精氨酸血管加压素;AT-2:血管紧张素-2;ACTH:促肾上腺皮质素;LH:黄体生成素;FSH:卵泡生成素;GnRH:促性腺激素释放激素
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
2.急性应激反应
应激能明显促进中枢多种神经递质的释放,其中最显著的是去甲肾上腺素和兴奋性氨基酸类。急性应激原能促进中枢尤其是下丘脑的去甲肾上腺素代谢和释放,但发生部位不尽相同,应激促进海马、前脑、皮质、杏仁核和下丘脑的去甲肾上腺素释放,组织中的浓度升高。在慢性应激时,接受了足够强度的应激原刺激后,脑组织对新的应激原敏感性增加;再次应激时,去甲肾上腺素的释放显著高于初次应激反应,称为“应激性去甲肾上腺素增敏”(stress-induced sensitization of norepinephrine)现象。 “应激性高血压”是一种生理反应而非病理变化,主要与心排血量增加、心率加快(伴外周血管阻力增加)以及儿茶酚胺、皮质醇、AVP、内啡肽和醛固酮分泌增多等有关[5]。如机体长期处于慢性应激状态,可发生慢性高血压(原发性高血压),血管壁增生、肥厚及动脉硬化。但一些继发性高血压可能主要是血管本身病变所致,如肝和肾等器官移植术后,患者常发生移植后高血压,这种高血压可能主要与环孢素诱导血管内皮细胞功能紊乱及细胞因子分泌有关,一氧化氮(NO)系统的活动被上调,而环孢素诱导的过氧化物和自由基可拮抗血管扩张导致高血压。此外,器官移植术后高血压也与血容量增多,交感-肾上腺皮质兴奋和应用糖皮质激素等有关。应激系统除激发觉醒和维持生命体征外,应激时中枢神经系统的皮质边缘多巴胺能系统、杏仁核海马复合体和弓状核POMC神经系统均被激活。
(1)皮质边缘系统:
属于多巴胺系统,由LC/NE-交感系统支配,对认知功能、欣快或抑郁心理状态有影响。
(2)杏仁核/海马复合体:
由脑干儿茶酚胺能神经元的神经纤维传入。杏仁核对任何应激情绪有重要作用。
(3)阿黑皮素原(POMC):
疼痛感觉由LC/NC-去甲肾上腺素能和CRH/AVP神经调节。α-MSH和β-内啡肽能抑制应激系统中枢的LC/NE和CRF的信息,投射到脑干和脊髓,产生镇痛作用。
(4)体温和食欲调节:
激活LC/NE-去甲肾上腺素和PVN,CRH系统使体温中枢升高体温。CRH对IL-1、TNF-α和IL-6所介导的部分产热作用是由外源性热源脂多糖(细菌的产物)的刺激所产生的。食欲和饮食中枢位于下丘脑内。CRH和α-MSH是一个主要的厌食性神经肽,它接受神经肽Y的刺激,而NPY却是一个较强的食欲性神经肽。在刺激CRH的同时,NPY还抑制LC/NE-交感系统和副交感神经系统的作用,帮助消化和储存营养。由脂肪细胞所分泌的瘦素是NPY的抑制剂。这些因子在应激系统激活状态下调节食欲。糖皮质激素也可刺激下丘脑NPY基因的表达,并且同时抑制PVN、CRH和LC/NE交感系统。
(三)急性应激性高血糖症
由于应激时代谢消耗的需要,机体动员葡萄糖调节机制,升高血糖,但有时由于各种原因或血糖调节机制障碍,亦可出现应激性低血糖症。在儿科急性疾病患者中,约占4%~5%。应激时,由于一些抗胰岛素激素(胰高血糖素、肾上腺素、GH和皮质醇等)分泌增多和胰岛素分泌受抑制所致。血糖升高程度不一,如血糖调节机制正常,不会引起持续性高血糖,但如原有糖耐量异常(IGT)可加重病情并出现糖尿病;如原患有糖尿病者,可诱发高渗性非酮症性昏迷或酮症酸中毒。因此,凡遇有急、慢性应激者,均不宜作OGTT检查(假阳性),但应进行常规血糖测定。
应激性高血糖是应激反应的评价指标之一,长期存在的高血糖和糖耐量减退(病前正常)提示机体应激反应没有解除;另一方面,应激性高血糖是心血管疾病的危险因素之一,研究表明,与急性心肌梗死相关的高血糖明显增加充血性心衰和心源性休克的危险性[13]。心肌梗死的病死率也明显高于无高血糖者。对糖尿病患者来说,应激性高血糖增加了氧化应激的严重程度,氧自由基生成增多引起胰岛素抵抗、糖利用减少及细胞的各种损害。形成胰岛素抵抗和氧自由基间的恶性循环,而高血糖是该恶性循环的始动和促发因素。慢性应激和糖尿病时,高血糖氧化应激诱导细胞间黏附分子-1(ICAM-1)表达,促进动脉粥样硬化形成。此外,1型糖尿病患者由于内源性吗啡样肽的反应减退症又减弱了机体的应激防御能力。应激时,增高的糖皮质激素不仅抑制GH和类固醇性激素的产生,还拮抗这些激素在脂肪组织中的分解代谢作用及肌肉和骨骼中的合成代谢。抑制成骨细胞的活性,产生骨质疏松。糖皮质激素又可导致胰岛素抵抗,因此各种应激激活HPA轴后,会使糖尿病的血糖不易控制。糖皮质激素引起的进行性腹部脂肪增加会直接加重2型糖尿病患者的胰岛素抵抗,产生高血糖和高胆固醇血症,还可能引发神经性病变等。
(四)应激性高血糖并发症
急性应激时,患者往往不能进食或因为病情和治疗需要被迫禁食,必须由静脉补充足够的糖类,但应激患者宜尽早恢复饮食,减少肠外营养支持量和时间。如因病情需较长时间应用静脉营养,必须考虑高血糖带来的毒性作用[13-15]。此外,长期静脉输入葡萄糖液也易发生葡萄糖毒性作用。急性高血糖的毒性作用主要表现为脱水和高渗状态;慢性毒性作用则指慢性高血糖状态对机体代谢和免疫的影响。高血糖的主要影响:①产生急性氧化应激;②胰岛素合成减少,出胞障碍,Ⅰ相分泌减弱或缺乏;③糖分子亲水,高渗糖液使血容量增加,心肺负荷加重;④糖的渗透性利尿作用可直接损害肾小管上皮细胞,使肾小管的重吸收和分泌功能降低,无机盐丢失;⑤糖进入肝脏,导致脂肪与氨基酸代谢障碍,体内成糖成酯作用消耗胰岛素,生成过多三酰甘油,使肝脏解毒功能下降;⑥干扰正常免疫功能,致细胞免疫功能下降;⑦损害微血管;⑧损害大血管,引起大动脉粥样硬化;⑨糖化蛋白形成(HbA1c、清蛋白、球蛋白、抗体、膜蛋白、转运蛋白和酶等)和糖化脂类形成(糖化IDL、糖化VLDL、糖化LDL和糖化HDL等),干扰细胞的正常代谢过程;⑩胰岛β细胞损害。慢性高血糖干扰凝血和纤溶过程,形成高凝状态;如肺水肿、脑水肿、肺炎、肾炎、肝炎、心肌炎和浆膜积液等疾病伴有高血糖,除加重原有病情外还可诱发或加重感染。
血糖对糖利用的影响遵循“质量作用定律”原理。不依赖于胰岛素但有赖于糖转运蛋白(GLUT)的携带,胰岛素促进GLUT4的“转位”,以加强糖利用。正常情况下,约50%的糖利用是通过质量作用原理来完成的。1型和2型糖尿病的空腹高血糖状态从组织糖利用的角度来看,代偿了胰岛素抵抗或缺乏所致的糖利用不足,但代偿不能或不全(部分代偿)。而糖的浓度效应减弱,这就是所谓的“葡萄糖抵抗”或“葡萄糖中毒”现象。肌肉和脂肪组织既是胰岛素抵抗也是葡萄糖抵抗的主要组织。骨骼肌遇有氧化应激时,出现胰岛素抵抗现象,肌细胞在接触H2O2时,葡萄糖的转运和糖原合成被抑制,蛋白激酶被激活。激活后酶的活性为通常情况的8~38倍,其中p38-MAPK的活化是导致胰岛素抵抗的最主要因素。葡萄糖抵抗的原因未明,可能与GLUT的降调节、糖氧化通路阻滞、非酯化脂肪酸(NEFA)升高并优先氧化以及无氧氧化过多等有关。另外,许多氨基酸和嘌呤代谢中间物或终末产物的堆积均可使糖氧化酶失活。正常人输注大量葡萄糖液后血糖可呈短时升高,一般不会出现毒性作用,但老年人,心、肾、脑功能不全及有IGT或糖尿病者则可发生水中毒、急性心衰、肺水肿和脑水肿等并发症。2型糖尿病(尤其是老年患者)存在明显的氧化应激反应,血中氧化型谷胱甘肽浓度升高,还原型和氧化型谷胱甘肽比值下降,血维生素C降低,单核细胞和粒细胞生成的NO增多,如再做OGTT试验,上述变化加剧。
应用高渗性葡萄糖液诱发的高渗性非酮症性昏迷病例屡有报道。应激时,由于拮抗胰岛素作用的应激激素分泌增多,加上老年人、孕妇或原有糖代谢紊乱者的失水和渴感中枢敏感性下降等原因,可于数小时至数日内发生高渗性昏迷,其病死率高。因此凡重症患者必须监测血糖和血(尿)渗透压等指标。慢性高血糖症是导致糖尿病一系列慢性并发症的重要原因,大血管和微血管长期处于“葡萄糖中毒”、氧化应激和免疫失调状态,高血糖可通过自由基损害血管壁、肾系膜细胞、视网膜细胞和神经纤维及胰岛β细胞,导致各种慢性并发症。从细胞水平和分子水平来看,这些病理变化均与氧化应激有直接或间接联系。
(五)急性应激与低血糖症
除药物(包括胰岛素制剂)外,应激因素是常见的低血糖症病因,其中以肝源性、心源性和肾源性低血糖症更常见。如进食过少、消耗过多、拮抗胰岛素激素缺乏或肝糖原和肌糖原被消耗后,易发生低血糖症。禁食或餐后血糖的重要来源是肝糖原分解和肝糖异生,低血糖伴血乳酸增加提示糖异生障碍。肝功能严重损害时常伴发严重的空腹低血糖症,慢性广泛性肝损害引起低血糖的另一原因是肝脏灭活胰岛素的能力下降及胰岛分泌的胰岛素由侧支循环大量进入体循环,表现为胰岛素/C肽比值明显升高。心源性低血糖症较少见,可能与肝淤血、心源性肝硬化、进食过少和肝糖异生障碍等有关。肾源性低血糖症主要与葡萄糖转换降低、丙氨酸成糖作用减弱以及肾灭活胰岛素减少等有关,部分患者的血丙氨酸降低。
(一)精神性身材矮小
应激时,HPA轴抑制GH的分泌和IGF-1生成,可能是由于某些配体与糖皮质激素受体结合,抑制c-jun/c-fos二聚化和基因转录所致。应激反应开始时,血GH浓度升高,这是通过糖皮质激素反应元件刺激GH基因转录所致;当应激时间延长和糖皮质激素持续增高时,即直接抑制了生长中枢。在发生烦躁或抑郁等情绪或心理的异常情况下,应激系统HPA轴处于慢性应激状态,血GH和IGF-1显著减低,这表明情绪抑郁引起的精神性矮身材是由于应激抑制GH分泌减低的缘故。
(二)肥胖或消瘦
慢性应激减低食欲,慢性应激时发生肥胖还与应激影响其胃肠功能有关,应激影响消化功能的主要作用部位在中枢神经的CRH分泌,PVN和CRH抑制胃酸的分泌和胃的排空,同时刺激结肠蠕动,使排便增加。腹部手术后,CRH分泌增加或由于中枢性IL-1水平增高抑制胃蠕动,排便的次数增多,并有腹痛(肠应激综合征)。
(三)免疫功能降低
感染性疾病激活自身免疫过程和HPA轴。炎症产生的TNF-α、IL-6和IL-1可激活HPA轴。炎性介导物包括花生四烯酸类(eicosanoid)、血小板激活因子和上皮生长因子等。另一方面,激活的HPA轴对免疫炎性反应有明显抑制作用,因为所有的免疫反应成分均被皮质醇所抑制。皮质醇的抗炎和免疫抑制作用降低白细胞的功能,使细胞因子和炎性介导物的产生都减少。
无论在急性、亚急性或慢性应激时,均表现为机体各种免疫功能减退,易并发感染和诱发各种躯体-精神性疾病:①应激是各种传染病、消化性溃疡、冠心病、高血压、肿瘤、格雷夫斯病、库欣综合征和精神分裂症等疾病的重要诱因或病因。例如,大面积烧伤者常并发柯林溃疡(Curling ulcer)和应激性心脏猝死等;②淋巴细胞增殖力下降,淋巴因子分泌减少,自然杀伤(NK)细胞功能障碍,外周血白细胞总数下降,特异性抗体生成障碍等;③免疫稳定功能紊乱,免疫功能下降时,有些致病性免疫因子或免疫抗体增多,免疫稳定和抗病机制失去正常平衡,如持续存在,易导致组织器官损伤和免疫性疾病,甚至肿瘤。应激性免疫功能紊乱机制未明。大量动物实验和临床观察证实,机体免疫功能紊乱与某些内分泌激素的过度分泌有关。激素对机体免疫功能的作用,见表1。
表1激素对机体免疫功能的作用
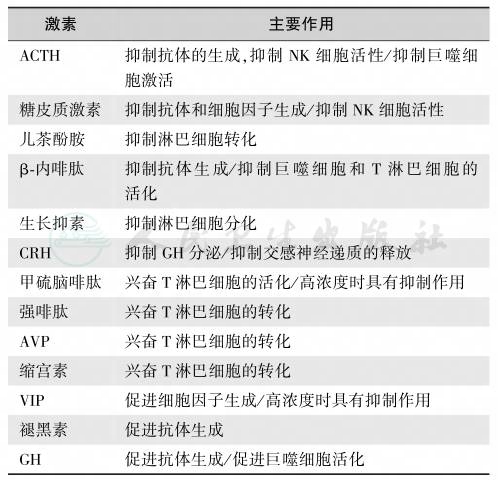
注:ACTH:促肾上腺皮质激素;CRH:促肾上腺皮质激素释放激素;AVP:精氨酸血管加压素;VIP:血管活性肠肽;GH:生长激素
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
由上表可知,参与免疫功能调节的激素很多。对某种免疫功能或免疫物质来说,均存在兴奋和抑制两方面激素的调节,但在应激状态下,以抑制免疫功能的激素占优势。此外,应激时机体产生一类免疫抑制因子。许多激素、细胞因子和免疫因子均有抑制免疫和增强氧化应激功能。因此,应激性免疫功能紊乱是神经内分泌、局部激素与细胞因子调节障碍所致,与自由基的过氧化作用(见下述)有关。在局部组织,免疫细胞还合成和分泌旁分泌与自分泌激素,参与免疫调节或作为免疫反应的应答。这些由免疫细胞产生的局部激素在免疫病理反应中起着十分重要的作用,当生成量过多时,可损伤靶组织。
(四)细胞衰老
慢性应激通过行为和生物途径引起病变。人们面临的社会存在强烈的心理应激和进食过多,而且心理应激与进食过多两种因素相互作用,进一步强化慢性应激过程[16-18]。慢性应激促进进食,升高皮质醇和胰岛素水平,抑制代谢分解激素的释放(代谢应激),导致腹部脂肪积聚、肥胖、系统性炎症和氧化应激。在这种生化环境下,通过细胞老化机制端粒末端转移酶(telomerase)被抑制,端粒长度(telomere length,TL)缩短,细胞衰老。免疫细胞端粒缩短引起许多疾病状态,导致各种并发症(图2)。
在细胞分裂过程中,由于DNA聚合酶功能障碍而不能完全复制染色体,复制的DNA序列可能会丢失,最终造成细胞衰老死亡。端粒是真核生物染色体末端由许多简单重复序列和相关蛋白组成的复合结构,这种保护性核蛋白结构具有维持染色体结构完整性作用。端粒酶是一种反转录酶,由RNA和蛋白质组成;以自身RNA为模板,合成端粒重复序列,加到新合成DNA链的末端上。大多数的胚胎组织、生殖细胞、炎性细胞、增生细胞及肿瘤细胞存在端粒酶。细胞有丝分裂后,有一段端粒序列丢失,当端粒长度缩短到一定程度时,细胞停止分裂,导致衰老与死亡。
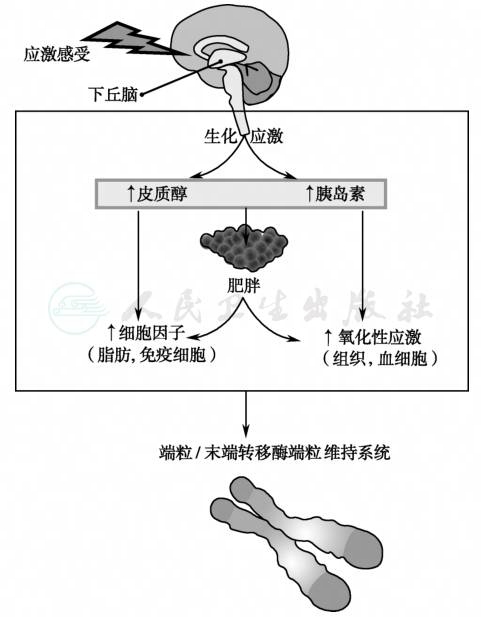
图2慢性应激作用
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
端粒/端粒酶活性与细胞衰老有一定联系。真核生物的体细胞端粒酶活性受抑制;精子的端粒要比体细胞长,体细胞缺失端粒酶活性后逐渐衰老,而生殖细胞系的端粒却可以维持其长度,而转化细胞能通过端粒酶完全复制端粒,维持其永生特性。但是,体细胞端粒长度与个体的寿命及不同组织器官的预期寿命并非一致。不同体细胞的有丝分裂能力不尽相同,胃肠黏膜细胞的分裂增殖速度快,神经细胞分裂的速度慢。鼠的端粒比人类长5~10倍,寿命却比人类短,生殖细胞端粒酶活性高,却不会无限制地分裂繁殖。
(五)代谢综合征
过度营养引起血糖、游离脂肪酸和氨基酸升高,诱导中枢性代谢性炎症;这些营养物质进入细胞内,线粒体和内质网在氧化营养物质和合成蛋白质时引起应激反应[19,20]。细胞内的反应性氧族增加,引起细胞内氧化应激。在此种环境中,细胞的高代谢状态需要内质网合成更多的蛋白质,因此导致内质网应激和细胞自噬。上述的线粒体应激、内质网应激、细胞自噬和炎症反应激活前炎症性激酶,如 IκB 激酶(IKK)和 c-Jun N 末端激酶(JNK),炎性细胞因子损害细胞结构和功能,引起能量代谢调节障碍和代谢综合征(图3)。
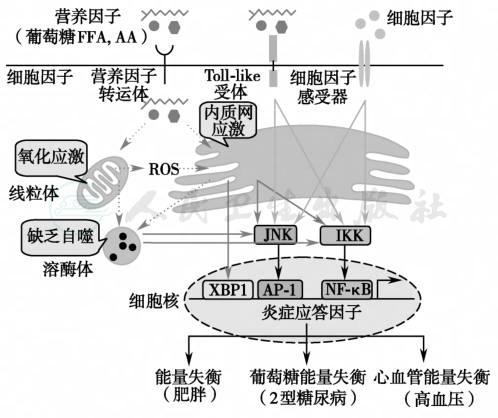
图3脑应激与代谢综合征
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
(六)细胞凋亡
自由基是具有非配对电子的基团或分子,正常机体及病理过程中均存在,生物体内多种物质代谢均可产生活性氧。活性氧包括超氧阴离子(O-2)、过氧化氢(H2O2)、羟自由基(—OH)及单线态氧(1O2)等。研究表明,H2O2、活性氧中间产物(ROI)、氧化应激、电离辐射和紫外线照射均能诱导细胞凋亡,可能由于它们产生了H2O2和—OH等活性氧,导致氧化应激而致细胞损害。另外,化疗和γ-干扰素等诱导细胞凋亡也与自由基的产生有关。暴露于低剂量的 H2O2(10~100μmol/L)能诱导多种细胞凋亡。氧化应激诱导细胞凋亡的机制可能与活性氧中间产物等自由基对DNA损伤,导致核糖转移酶活化和p53的积累以及脂质过氧化物使细胞内Ca2+增加有关。
热损伤诱导热休克反应。热休克蛋白(HSP)作为一类在进化过程中具有高度保守性的应激蛋白,对维持机体的自身稳定性起着重要作用。HSP可分为 HSP110、HSP90、HSP70、HSP60、HSP47、HSP32、HSP28、HSP10 及 ubiquitin(泛激素)等数种。NO可诱导HSP70表达,从而阻止TNF-α诱导的细胞凋亡。另外,五羟黄酮(quercetin)可导致肿瘤细胞凋亡并伴HSP70减少,认为可能是通过抑制HSP70的产生而导致肿瘤细胞凋亡。
脑缺血再灌注损伤中存在迟发性神经细胞死亡(DND),脑组织的各部位细胞对缺血的敏感性不同,如海马、纹状体及皮质区的神经元为易受暂时缺血损伤的部位。一过性脑缺血后,海马CAI区域和纹状体背外侧和海马区域细胞出现凋亡,大脑缺血再灌注时,由于炎性细胞的浸润及氧的内流而引起氧自由基增加,进而引发细胞凋亡。
(七)细胞坏死与慢性增殖性炎症
应激刺激通过IL-1β和TNF-α,或由于缺血损伤、热休克、低渗状态和机械牵张等原因,胞内SAPK被活化,调控转录因子活性改变影响基因表达,从而使细胞产生不同的应激性生物效应。另一种ERK家族成员p38在应激时也可被活化,其作用类似于SAPK。缺血本身激活 SAPK,如肾动脉缺血后再灌注时SAPK迅速被激活,使细胞产生适应性反应。严重缺血时细胞可发生坏死,但缺血不严重而细胞又分化不完全时,细胞则停止分化进入循环周期,以替代那些不可逆损伤的细胞。SAPK和p38活化能促进细胞进入循环周期或调控生长阻抑状态,使细胞对缺血再灌注损伤产生适应,获得幸存。
动脉粥样硬化(AS)斑块和狭窄的新内膜形成是一种特殊的增殖性慢性炎症,在这些病理过程中,SAPK调控细胞基因表达(尤其是细胞增殖相关基因的表达),形成细胞增殖性病变;其中诱导基质金属蛋白酶(MMP)表达是激酶活化的重要标志。MMP活化后引起细胞外基质降解,这是AS斑块纤维帽变薄甚至破裂的重要原因。SAPK活化后可引起AP-1激活,后者调控金属蛋白酶酶基因启动子,因此在AS斑块形成与破裂、狭窄病变和平滑肌细胞增殖后的迁移等过程中,SAPK的参与起着关键作用。
氧化应激是生物体内的抑制正常现象。正常情况下,细胞内的氧化还原系统使活性氧(氧自由基,ROS)和活性氮(氮自由基,RNS)维持在低水平,因此,氧化应激是机体氧化剂与抗氧化剂不平衡的一种表现。活性氧与活性氮过多引起氧化应激性疾病,如动脉粥样硬化、高血压、心肌梗死、糖尿病、慢性疲劳、自身免疫性疾病、黄斑变性、神经变性性疾病、哮喘、类风湿与骨关节病、肾炎等。
(一)应激反应
应激是由精神心理环境因素引起的体内生化反应过程[26],激活神经激素调节机制并影响机体的生长、发育生殖等功能[27-29],但只要应激原消除,应激反应即停止,恢复原来的生理水平[30-32]。如果机体的敏感性过高或已经反应过强即可导致病理性应激反应和疾病。应激时身体的表现多样[33]。导致氧化应激的应激原称为促氧化剂(prooxidant),常见的促氧化剂有寒冷、体力活动、慢性应激、营养性应激、缺氧性应激等,见图4。
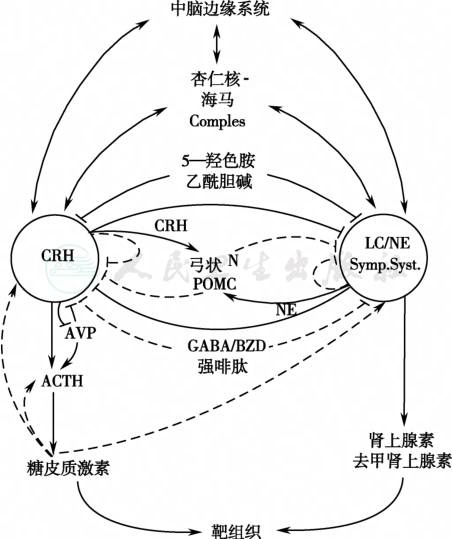
图4应激反应系统
下丘脑-垂体-肾上腺轴和去甲肾上腺素交感系统相互作用组成应激反应系统
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
(二)应激生化反应
一些细胞和细胞组分参与了ROS的生成反应的启动与扩增[34-37],见表2。下丘脑-垂体-肾上腺轴与下丘脑-垂体-性腺轴的相互作用特点是肾上腺被兴奋而性腺功能被抑制(图5),同时下丘脑-垂体-肾上腺髓质去甲肾上腺素交感系统被激活,儿茶酚胺分泌增多(图6和图7)。
表2氧化应激的内源性介导物
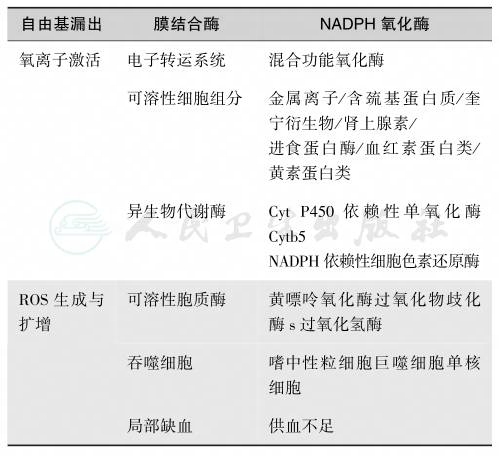
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
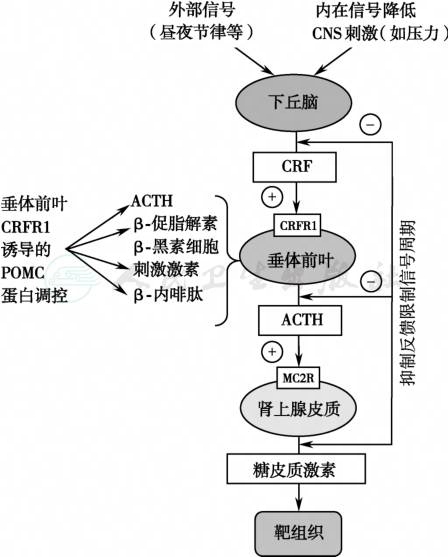
图5下丘脑-垂体-肾上腺轴应激反应
HPA轴是机体应激的主要反应系统;应激时下丘脑释放CRF,通过血液进入垂体,与其受体CRFR1结合后激活CRFR1,促进POMC分解和ACTH生成;ACTH在肾上腺与MC2R结合,刺激糖皮质激素(人类主要是皮质醇,动物主要是皮质酮)释放;而糖皮质激素反馈抑制CRF和ACTH释放
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
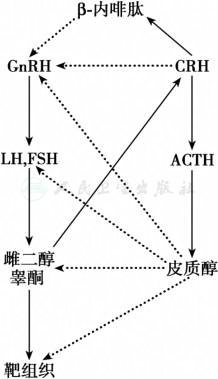
图6下丘脑-垂体-肾上腺轴与下丘脑-垂体-性腺轴的相互作用
应激系统激活通过CRH、β-内啡肽和糖皮质激素抑制下丘脑-垂体-性腺轴功能
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
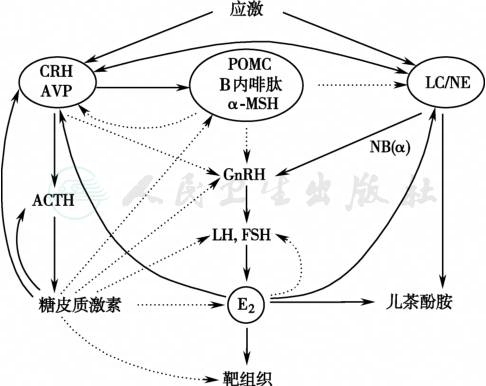
图7下丘脑-垂体-肾上腺轴与去甲肾上腺素交感系统及下丘脑-垂体-性腺轴的相互作用
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
(三)氧化应激与疾病
活性氧(ROS)和活性氮(RNS)自由基引起生物学损害的现象分别称为氧化应激和硝化应激[38-40],其原因是ROS/RNS过多或抗氧化剂缺乏。导致病变的严重性与氧化物-抗氧化剂的性质、含量、病变部位与强度以及机体的修复能力有关。ROS包括分子氧的所有代谢产物,这些物质包括超氧化物自由基(superoxide radical,O2•)、羟自由基(hydroxyl radical,HO•)和非自由基分子、H2O2。它们的化学反应能力远高于氧分子(O2),可引起各种慢性病变(表3)[41-46]。
表3与氧化应激密切相关的疾病
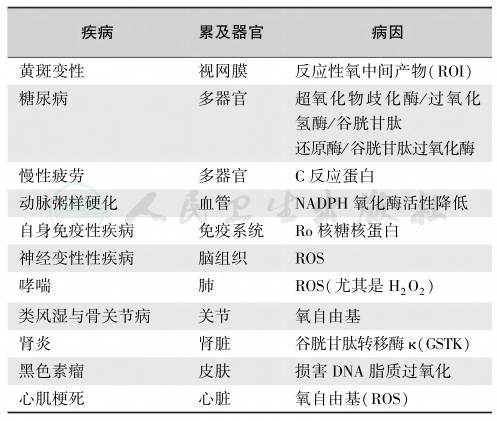
注:ROI:反应性氧中间产物(reactive oxygen intermediates);GSTK1-1:谷胱甘肽转移酶 κ(glutathione transferase)
引自:内分泌代谢病学(全2册).第4版.ISBN:978-7-117-27841-6
(四)线粒体应激与疾病
在应激情况下,细胞受到刺激后,IP3和cADP核糖(cADPr)从肌质网通过其受体-P3受体-里阿诺碱受体,IP3R和兰尼碱受体(ryanodine receptor,RyR)释放出来。升高的细胞质Ca2+被线粒体钙单向转运体(calcium uniporter,CaU)摄取,由此改变了 Ca2+/H+交换体(非 Na+依赖性 Ca2+交换体,Na+-independent Ca2+exchanger,NICE,CHE)的功能。线粒体内 Ca2+激活还原型底物(NADH),电子转运链的电子转运加速,ROS生成增多。后者刺激IP3R和RyR,促进Ca2+进一步释放。线粒体呼吸链功能亢进使质子泵活跃,CHE被进一步激化,使更多的Ca2+进入线粒体内,引起线粒体应激。
引起线粒体应激的因素很多,如高钠饮食、低钙饮食、低维生素D饮食、各种慢性应激状态、慢性高肾上腺素性应激状态、高血压、原发性高血压、低肾素性高血压、盐敏感性高血压、原发性醛固酮增多症、慢性充血性心衰或慢性肾衰等。
线粒体应激时,血浆和细胞外液Ca2+降低而细胞内Ca2+积蓄,及形成无症状性继发性甲状旁腺功能亢进症,继而心肌线粒体离子钙过负荷和细胞器氧化应激,线粒体溶质性肿胀与变性。在心脏,出现心肌坏死-纤维化及心衰。此外,细胞外Ca2+和Zn2+通过L型Ca2+通道(LTCC)进入细胞内,Ca2+作为促氧化剂而Zn2+作为抗氧化剂发挥作用。细胞质[Ca2+]i和[Zn2+]i以及线粒体[Ca2+]m与[Zn2+]m增高,Ca2+过负荷诱导氧化应激,上调的Zn2+转运进一步促进Zn2+进入细胞内。释放的无活性Zn2+与结合蛋白——金属硫蛋白-1(MT-1)结合。[Zn2+]i与[Zn2+]m进一步升高,心肌细胞促氧化剂与抗氧化剂失衡引起细胞凋亡与纤维化。慢性心衰既是线粒体氧化应激的后果,也可以是其原因,例如慢性心衰并发继发性甲状旁腺功能亢进症的病因包括维生素D缺乏、高醛固酮血症和髓袢利尿剂,而高PTH血症恶化心血管功能的途径包括Ca2+过负荷、能量贮存过多、线粒体功能紊乱和细胞因子分泌增多等。
1.TachèY.Hans selye and the stress response:from “the first mediator”to the identification of the hypothalamic corticotropin-releasing factor.Ideggyogy Sz,2014,67(3-4):95-98.
2.Somogyi A.Sslye's concept of pluricausal diseases and its impact on regulatory science.Ideggyogy Sz,2014,67(3-4):87-90.
3.Kubo KY,Iinuma M,Chen H.Mastication as a Stress-Coping Behavior.Biomed Res Int,2015,2015:876409.
4.Ono Y,Yamamoto T,Kubo KY,et al.Occlusion and brain function:mastication as a prevention of cognitive dysfunction.J Oral Rehabil,2010,37(8):624-640.
5.Furtado M,Katzman MA.Neuroinflammatory pathways in anxiety,posttraumatic stress, and obsessive compulsive disorders.Psychiatry Res,2015,229(1-2):37-48.
6.Briere J,Scott C.Complex trauma in adolescents and adults:effects and treatment.Psychiatr Clin North Am,2015,38(3):515-527.
7.Juskewitch JE,Huskins WC.Reply:evaluation of respiration and heart rate decreases reliability of the pediatric systemic inflammatory response syndrome definition.Pediatr Crit Care Med,2012,13(3):371-372.
8.Zurek J,Vavrina M.Procalcitonin biomarker kinetics to predict multiorgan dysfunction syndrome in children with sepsis and systemic inflammatory response syndrome.Iran J Pediatr,2015 Feb,25(1):e324.
9.Gharib SD,Berger DL,Choy G,et al.Caserecords of the massachusettsgeneralhospital.case 21-2015.A 37-year-old American man living in Vietnam,with fever and bacteremia.N Engl J Med,2015,373(2):174-183.
10.Nogueira C,Borges F,Lameu E,et al.Retinol,β-carotene and oxidative stress in systemic inflammatory response syndrome.Rev Assoc Med Bras,2015,61(2):116-120.
11.Osborne DM,Pearson-Leary J,McNay EC.The neuroenergetics of stress hormones in the hippocampus and implications for memory.Front Neurosci,2015,9:164.
12.Elnakish MT,Ahmed AA,Mohler PJ,et al.Role of oxidative stress in thyroid hormone-induced cardiomyocyte hypertrophy and associated cardiac dysfunction: an undisclosed story.Oxid Med Cell Longev,2015,2015:854265.
13.Mukherjee JJ,Chatterjee PS,Saikia M,et al.Consensus recommendations for the management of hyperglycaemia in critically ill patients in the Indian setting.J Assoc Physicians India,2014,62(7 Suppl):16-25.
14.Fan X,Jiang Y,Yu Z,et al.Combination approaches to attenuate hemorrhagic transformation after tPA thrombolytic therapy in patients with poststroke hyperglycemia/diabetes.Adv Pharmacol, 2014, 71:391-410.
15.Korac'evic'G,Vasiljevic'S,Velickovic'-Radovanovic'R,et al.Stress hyperglycemia in acute myocardial infarction.Vojnosanit Pregl,2014,71(9):858-869.
16.Epel ES.Psychological and metabolic stress:a recipe for accelerated cellular aging? Hormones (Athens),2009,8(1):7-22.
17.Edifizi D,Schumacher B.Genome instability in development and aging:insights from nucleotideexcision repair in humans, mice, and worms.Biomolecules,2015,5(3):1855-1869.
18.Adnane B,Mainassara ZA,Mohamed F,et al.Physiological and molecular aspects of tolerance to environmentalconstraints in grain and forage legumes.Int J Mol Sci,2015,16(8):18976-19008.
19.Cai D,Liu T.Inflammatory cause of metabolic syndrome via brain stress and NF-κB.Aging (Albany NY),2012,4(2):98-115.
20.Taylor SR,Meadowcraft LM,Williamson B.Prevalence,pathophysiology,and management of androgen deficiency in men with metabolic syndrome,type 2 diabetes mellitus,or both.Pharmacotherapy,2015,35(8):780-792.
21.Henry JP.Biological basis of the stress response.Integr Physiol Behav Sci(Abstract),1992,27(1):66-83.
22.Kraemer WJ,Fleck SJ,MareshCM,et al.Acute hormonal responses to a single bout of heavy resistance exercise in trained power lifters and untrained men.Can J Appl Physiol,1999,24(6):524-527.
23.Hill BG,Ramana KV,Cai J,et al.Measurement and identification of S-glutathiolated proteins.Methods Enzymol,2010,473C:179-197.
24.Kovacic P,Thurn LA.Cardiovascular toxicity from the perspective of oxidative stress,electron transfer,and prevention by antioxidants.Curr Vasc Pharmacol,2005,3(2):107-117.
25.Rosen HR,Rich BA.Neurocognitive correlates of emotional stimulus processing in pediatric bipolar disorder: a review.Postgrad Med,2010,122(4):94-104.
26.Dimitrios NT,Geogrios KC,Dmitrios IXH.Neurohormonal hypothesis in heart failure.Hellenic J Cardiol,2003,44(3):195-205.
27.Lundgren K,Kalev K,Chuansi GA,et al.Effects of heat stress on working populations when facing climate change.Ind Health,2013,51(1):3-15.
28.Kock MD,Jessup DA,Clark RK,et al.Effects of capture on biological parameters in free-ranging bighorn sheep (Ovis canadensis):evaluation of drop-net,drive-net,chemical immobilization and the net-gun.J Wildl Dis,1987,23(4):641-651.
29.Hoult JR,Moroney MA,Paya M.Actions of flavonoids and coumarins on lipoxygenase and cyclooxygenase.Methods Enzymol,1994,234:443-454.
30.Akhalaya MY,Platonov AG,Baizhumanov AA.Short-term cold exposure improves antioxidant status and general resistance of animals.Bull Exp Biol Med,2006,141(1):26-29.
31.Kock MD,Clark RK,Franti CE,et al.Effects of capture on biological parameters in free-ranging bighorn sheep (Ovis canadensis):evaluation of normal,stressed and mortality outcomes and documentation of postcapture survival.J Wildl Dis,1987,23(4):652-662.
32.Ziegler R.Changes in lipid and carbohydrate metabolism during starvation in adult Manducasexta.J Comp Physiol B,1991,161(2):125-131.
33.Dhanalakshmi S,Srikumar R,Manikandan S,et al.Antioxidant property of triphala on cold stress induced oxidative stress in experimental rats.J Health Sci,2006,52(6):843-847.
34.Misra HP,Fridovich I.The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase.J Biol Chem,1972,247(10):3170-3175.
35.Yu BP.Cellular defenses against damage from reactive oxygen species.Physiol Rev,1994,74(1):139-162.
36.Stoian I,Oros A,Moldoveanu E.Apoptosis and free radicals.Biochem Mol Med,1996,59(2):93-97.
37.Younes M.Free radicals and reactive oxygen species,in toxicology,‘By H.Marguardt,Mechanisms of antioxidant and pro-oxidant effects of lipoic acid in the diabetic and nondiabetic kidney'.Kidney Int,1999,67(4):1371-1380.
38.Kovacic P,Jacintho JD.Mechanisms of carcinogenesis:focus on oxidative stress and electron transfer.Curr Med Chem,2001,8(7):773-796.
39.Ridnour LA,Isenberg JS,Espey MG,et al.Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1.Proc Natl Acad Sci U S A,2005,102(37):13147-13152.
40.Valko M,Morris H,Mazúr M,et al.Oxygen free radical generating mechanisms in the colon:do the semiquinones of vitamin K play a role in the aetiology of colon cancer? Biochim Biophys Acta,2001,1527(3):161-166.
41.Racek J,Treska V,Krizan V,et al.The significance of free radicals in operations of acute ischaemia of the limbs.Zeitschrift für klinische Chemie und klinische Biochemie,1995,3:103-105.
42.Pechan I,Danova K,Olejarova I,et al.Oxidative stress and antioxidant defense systems in patients after heart transplantation.Wiener Klinische Wochenschrift,2003,115(17-18):648-651.
43.Ballinger SW.Mitochondrial dysfunction in cardiovascular disease.Free Radic Biol Med,2005,38(10):1278-1295.
44.Hermann A,Sitdikova GF,Weiger TM.Oxidative stress and maxi calcium-activated potassium (BK) Channels.Biomolecules,2015,5(3):1870-1911.
45.Giorgio M.Oxidative stress and the unfulfilled promises of antioxidant agents.Ecancermedicalscience,2015,9:556.
46.Liu J,Wang Z.Increased oxidative stress as aselective anticancer therapy.Oxid Med Cell Longev,2015,2015:294303.









