


 收藏
收藏 已收藏
已收藏英文名称 :Carney complex
中文别名 :Swiss综合征;黏液瘤综合征;NAME综合征;LAMB综合征
Carney综合征(Carney complex,CNC)是一种特殊的MEN(MIM 160980),又名Swiss综合征、黏液瘤综合征、NAME综合征(nevi-atrial myxomas-myxoid-neurofibroma and ephelide)或LAMB综合征(lentigines-atrial myxoma-mucocutaneous myxomas-blue nevi)。由Carney于1985年首次完整而详尽地进行了描述,包括黏液瘤、皮肤黏膜斑点状色素沉着和内分泌功能亢进三项主要表现,各项也可单独存在,特别是心脏黏液瘤和色素沉着。由于该复合症患者常有两个或更多内分泌腺的肿瘤,因此被认为是多发性内分泌腺肿瘤综合征(MEN)中的一种特殊类型,多见于30~40岁的年轻人,白种人多见,女性明显多于男性,常有家族聚集性,约50%的患者有家族史。
(一)PPKAR1A突变
遗传性神经肿瘤综合征的发病机制见图1,神经纤维瘤蛋白的功能结构域及其细胞内信号途径和神经纤维瘤病的发病机制见图2和图3。merlin蛋白能与细胞核中的JunD/AP1、smad3、NF-κB和其他蛋白相互作用,对细胞转录和生长、DNA复制和修复均有调节作用。merlin蛋白抑制或激活转录,保持由转化生长因子B介导的信息转导(包括甲状旁腺和求偶素受体介导的转录),调节细胞的生长。merlin失活突变致使细胞生长失控而导致肿瘤的发生,其详细过程还需进一步研究。Merlin作为一种转录辅调节子或辅抑制子而影响募集组蛋白修饰酶的活性与组蛋白甲基化,后者通过募集混合谱系白血病蛋白的作用(图4和图5),NF-1可伴有恶性外周神经鞘瘤。Carney复合症的特点是伴有内分泌表现,而神经鞘瘤病只局限于外周神经组织内。
家系研究发现,该复合症符合常染色体显性遗传,致病基因位于2p16;分子遗传学研究发现该复合症与17q22-24区域相连锁,区域内cAMP依赖性蛋白激酶Aα调节亚基(PPKAR1A)基因突变被证实是导致CNC的原因。45%~65%的Carney复合症患者存在PRKAR1A杂合突变,而原发性色素性结节性肾上腺皮质病(PPAND)中PRKAR1A突变率高达80%。Carney复合症的发病机制和临床表现与家族性着色斑病综合征相似,后者主要包括McCune-Albright综合征(MAS,MIM 174800)、Peutz-Jeghers综合征、LEOPARD综合征、Noonan综合征、Cowden病和Bannayan-Ruvalcaba-Riley综合征,见图6。这些综合征的皮肤病变均伴有内分泌或其他异常。
Carney三联征(Carney triad)是指副神经节瘤(PGL)、胃肠间质细胞瘤(GIST)和肺软骨瘤(PCH)同时或异时地发生于同一个体的现象,有些患者可发生肾上腺皮质腺瘤、胃间质细胞瘤、食管平滑肌瘤、心脏黏液瘤、嗜铬细胞瘤、色素沉着性皮肤病变(约50%)等,因此Carney复合症是一种特殊类型的MEN。其中,色素沉着性皮肤病变在排除其他病因后,往往提示Carney复合症的诊断。
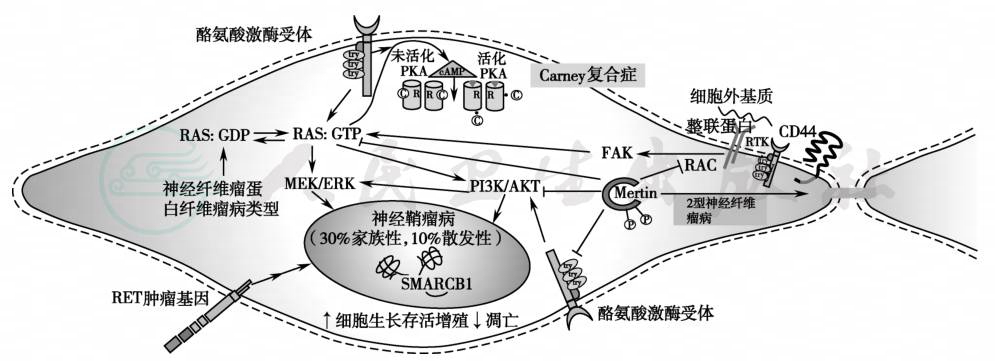
图1 遗传性神经肿瘤综合征的发病机制
神经鞘肿瘤的遗传易感性包括肿瘤抑制基因的胚系突变,神经纤维瘤蛋白(neurofibromin)功能缺失引起MEK/ERK信号通路活化和NF1,而NF2与merlin缺失有关,从而调节RAC、PI3K和受体酪氨酸激酶及PDGF受体功能异常;多数神经鞘瘤病呈散发性,SMARCB1基因编码SWI/SNF染色质重建复合物(chromatin remodeling complex),突变后导致神经鞘瘤病;PRKAR1A基因突变引起Carney复合症,MEN-2b由RET突变所致,一般不引起真正的外周神经肿瘤,但却促进错构瘤生长
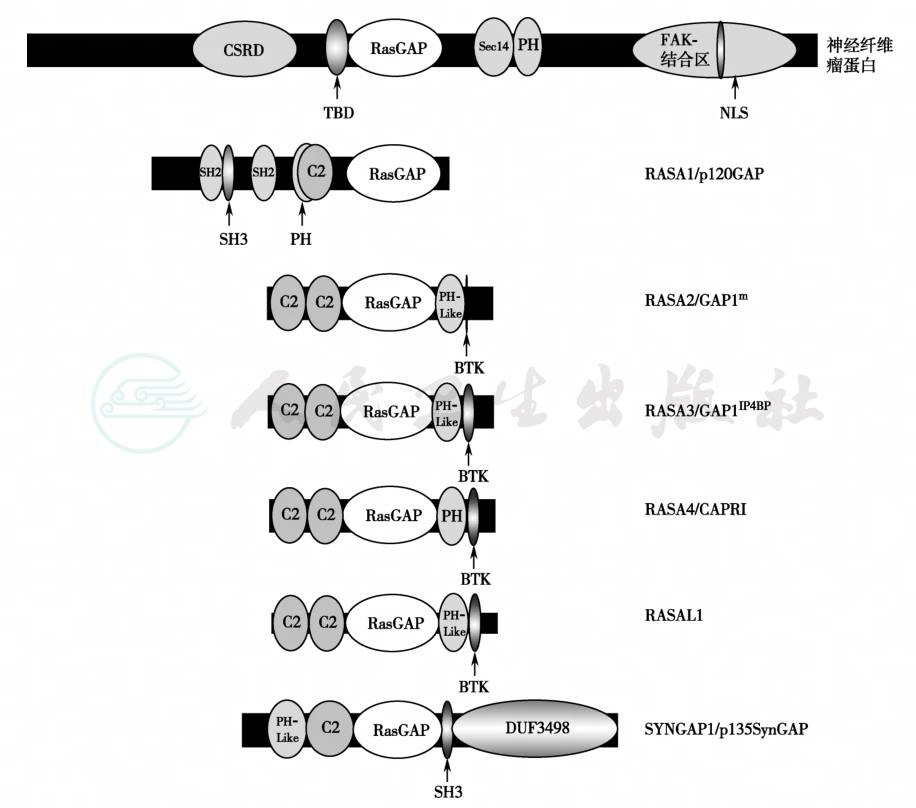
图2 神经纤维瘤蛋白的功能结构域
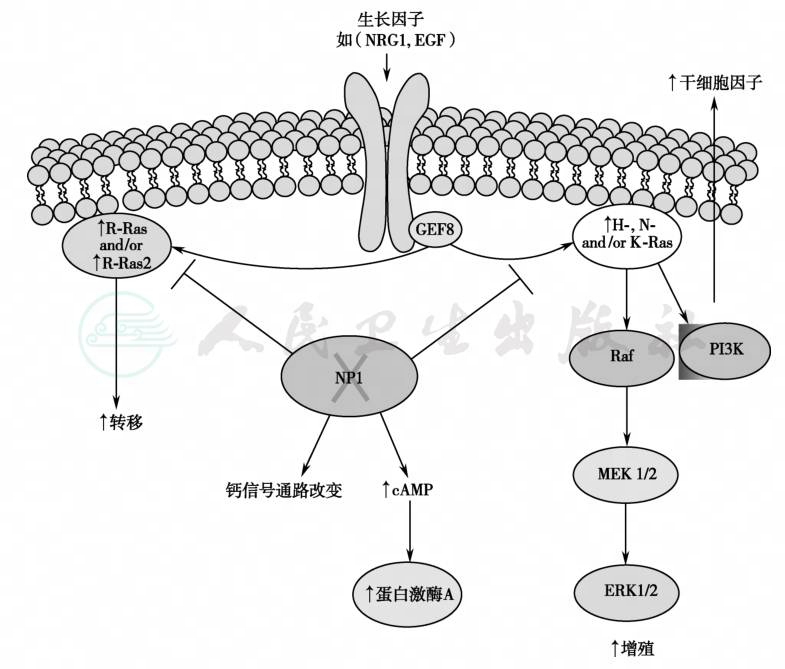
图3 神经纤维瘤蛋白的细胞内信号途径和神经纤维瘤病的发病机制
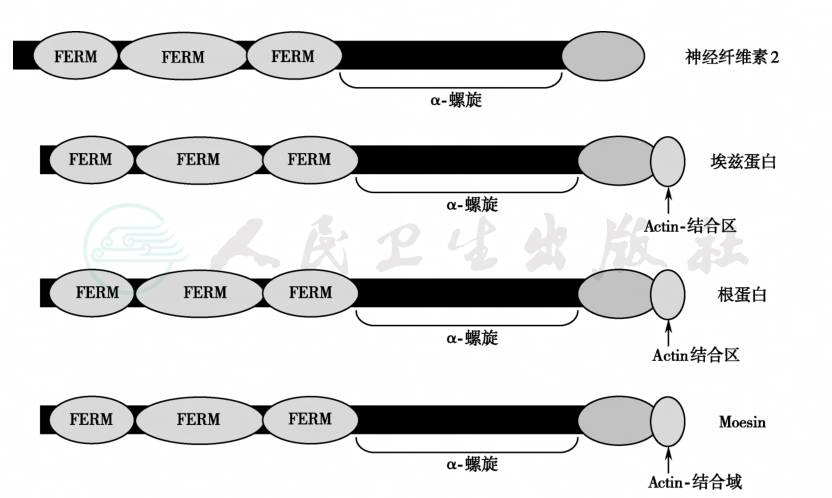
图4 merlin蛋白结构
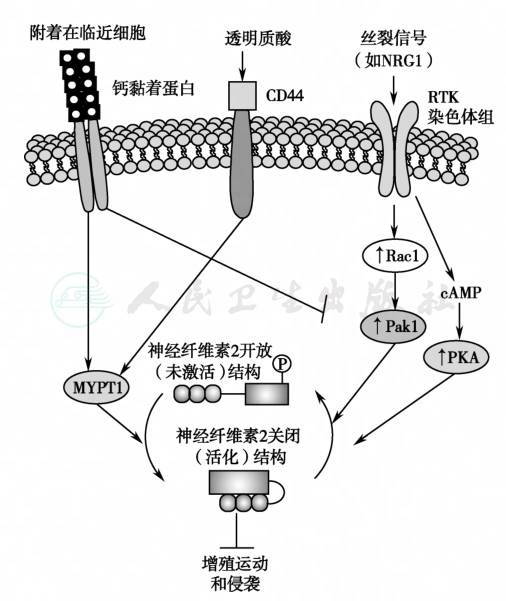
图5 merlin蛋白循环与信号途径
merlin去磷酸化被激活,磷酸化后被灭活;不同的跨膜受体激酶可激活或灭活merlin活性;RTK:受体酪氨酸激酶;PKA:蛋白激酶A;Pak:p21激活的激酶
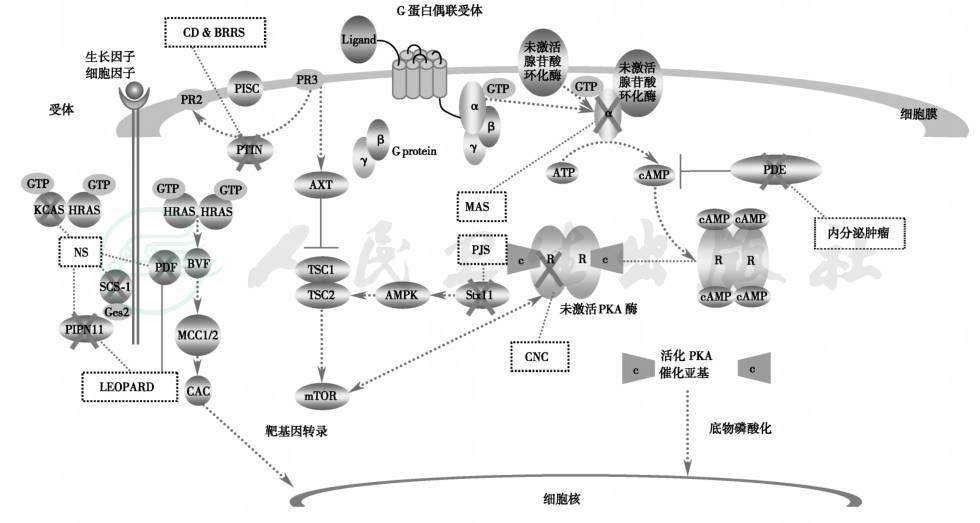
图6 家族性着色斑病综合征的发病机制
在家族性着色斑病综合征发病机制中,存在共同的分子病因基础,各种综合征之间又发生分子水平上的“信息对话”;CNC:Carney复合症;MAS:McCune Albright综合征;PJS:Peutz-Jeghers综合征;NS:Noonan综合征;CD:Cowden病;BRRS:Bannayan-Ruvalcaba-Riley 综合征;PIP2:磷脂酰肌醇-3,4,5-三磷酸;PIP3:磷脂酰肌醇-4,5-二磷酸;P13K:磷酸肌酸激酶;PTEN:磷酸酶与张力蛋白同源序列;TSC1/TSC2.hamartin/tuberin complex:错构瘤蛋白/抗结核菌素复合物;mTOR:雷帕霉素哺乳动物靶分子;AKT:v-akt murine thymoma viral oncogene homolog 1(protein kinase B),鼠胸腺瘤病毒原癌基因同源序列1(PKB);AMPK:蛋白激酶;AMP:活化的非催化亚基;KRAS:v-Ki-ras2 Kirsten鼠肉瘤病毒原癌基因同源序列;HRAS:v-Ha-ras Harvey鼠肉瘤病毒原癌基因同源序列;R:PKA调节亚基;C:PKA催化亚基
PPKAR1A基因突变导致细胞异常增殖,发生内分泌腺瘤。PRKAR1A是一种抑癌基因,编码PKA1α调节亚基蛋白。正常情况下,与蛋白激酶A(PKA)的催化亚基C构成四聚体(2个α调节亚基和2个C催化亚基)保持稳定。一旦PKA被上游信号激活,α亚基与cAMP结合并从催化亚基解离,使后者发挥催化活性,激活下游环腺苷酸反应元件结合蛋白(CREB)信号系统促进DNA复制,细胞生长和增殖。这2个α亚基分别由两条染色体编码。因此,当一条染色体上的基因发生突变使蛋白质合成异常时,缺少一个α亚基的PKA将无法保持其四聚体的稳定结构,导致PKA处于失抑制状态,下游信号被持续激活,最终导致细胞的异常生长增殖。CNC是临床上第1个被报道与PKA基因突变有关的疾病。因此,有学者将之称为“PKA病”。cAMP信号与内分泌肿瘤相关的最好例子是GNAS基因(Gsp癌基因,编码Gsa)突变引起的McCune-Albright综合征,GNAS活化性突变引起-腺苷酸环化酶被持续激活和PKA活化,出现多骨骼纤维增殖不良、café-au-lait皮肤斑和自发性内分泌功能亢进症。PRKAR1A基因突变引起的CNC也与PKA信号异常有关,cAMP信号通路与遗传性内分泌病关系见图7,Prkar1a与其他基因作用引起Carney复合症见图8。
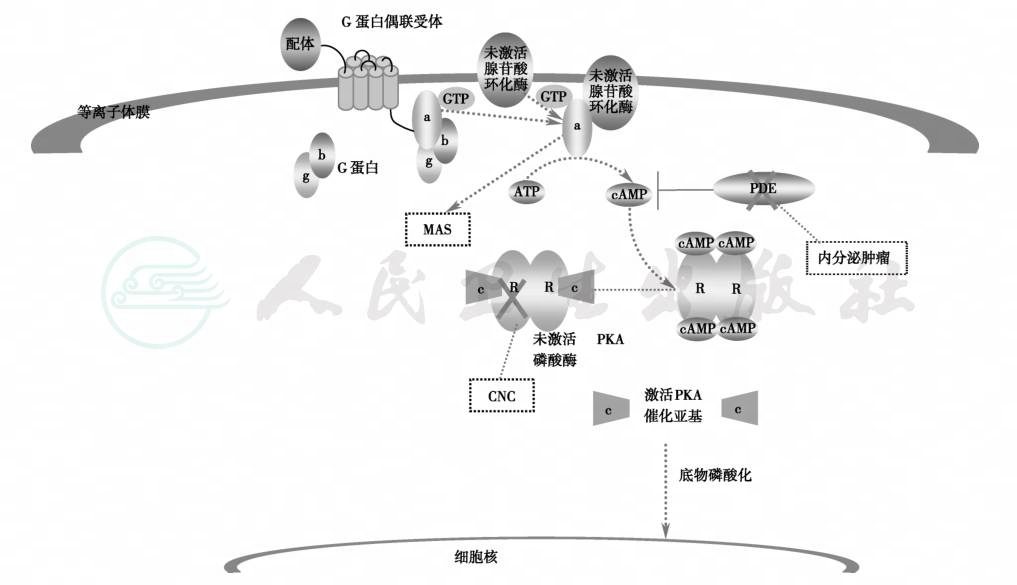
图7 cAMP信号通路与遗传性内分泌病
ATP:三磷酸腺苷;C:PKA 催化亚基,CNC:Carney复合症;GDP:二磷酸鸟苷;GTP:三磷酸鸟苷;MAS:McCune-Albright综合征;PDE:磷酸二酯酶;PKA:蛋白激酶 A;R:PKA 调节亚基;a/b/g:G 蛋白亚基 a/b/g
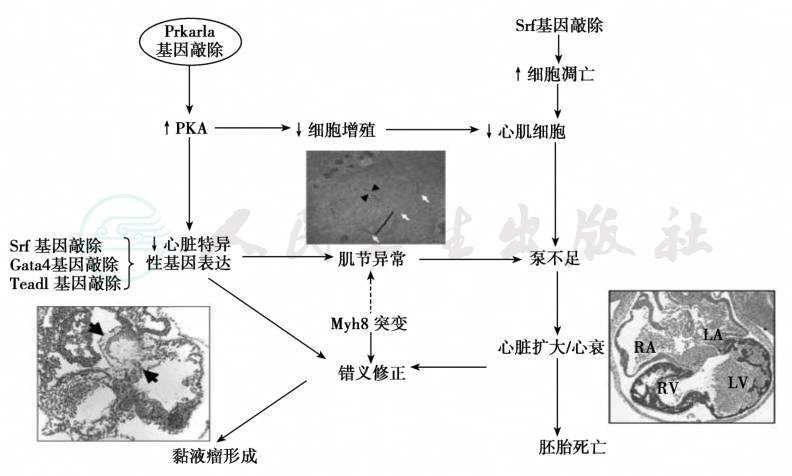
图8 Prkar1a与其他基因相互作用引起Carney复合症
Prkar1a基因敲除(Prkar1a KO)引起心脏特异性基因表达下降和心肌数目减少,肌节异常,Z-盘和I带残留,形成薄壁的扩张性心肌病(右侧)和黏液瘤形成(左侧);其他心脏特异性转录因子突变也引起同样病变
CNC相关的突变分布于PRKAR1A基因的10个外显子和少数内含子区域内,但以4B外显子较为集中,称为“热点”突变区域,可能是由于该区域的氨基酸序列也是PKA1α调节亚基与cAMP结合的关键结构域,功能易受基因突变的影响,而此区域最常见的突变是578delTG造成移码框突变而致mRNA转录提前终止。国内报道了4B外显子中一个新的点突变(S147N),认为可能是PPKAR1A的又一种突变型。PRKAR1A基因缺陷的外显率接近100%,但不累及垂体,而甲状腺和肾上腺肿瘤可发生PRKAR1A基因的体细胞突变。真核细胞的Wnt信号途径高度保守,控制着细胞的增殖与分化。CTNNB1基因的体细胞活化性突变是肾上腺皮质腺瘤、腺癌、PPNAD,Carney复合症的最常见分子事件,糖元合酶激酶3β是cAMP/PKA信号的下游靶分子,cAMP信号通过PKA及其转录因子CREB调节Wnt途径的基因表达。PPNAD和非ACTH依赖型大结节肾上腺增生的cAMP信号过表达,引起其靶基因WISP2、GSK3β和CTNNB1活性增高。
肾上腺皮质肿瘤与TP53基因的突变有关,首先可能出现良性增生和ADT克隆,继而可因Wnt信号异常、生长因子过表达和细胞周期蛋白异常(TP53,CHEK2)引起肾上腺癌或双侧肾上腺皮质增生性Cushing综合征。
(二)线粒体复合物Ⅱ琥珀酸脱氢酶突变
现已经证实,家族性和散发性副神经节瘤是由于线粒体复合物Ⅱ琥珀酸脱氢酶(SDH)失活性突变所致,SDH的3个亚基(SDHB、SDHC、SDHD)突变引起CNC,而c-kit(KIT)与血小板衍化生长因子受体A(PDGFRA)活化性突变导致家族性胃肠间质细胞瘤(GIST)。有些GIST患者伴有PGL,并呈常染色体显性遗传,即Carney-Stratakis综合征,亦可鉴定出种系性SDHB、SDHC和SDHD突变,而没有KIT或PDFGRA突变。因而,GIST的直接病因是SDH缺乏。确定Carney复合症患者GIST突变基因的意义在于:当GIST不存在cKIT和PDGFRA突变时,患者对酪氨酸激酶抑制剂是抵抗的,而当诊断为副神经节瘤时,则必须对其家族成员进行相关基因鉴定,早期发现和处理MEN。
错构瘤性息肉病综合征代表了另一类常染色体显性遗传性肿瘤综合征。患者特别容易发生结肠直肠癌和肠外肿瘤。CNC可人为地分为3类(表1),PRKAR1A突变(以c.491-492delTG常见)携带者和CNC患者均发生黏液瘤、斑点状皮肤色素沉着和内分泌功能亢进症。
表1 Carney复合症基因型与表型的关系
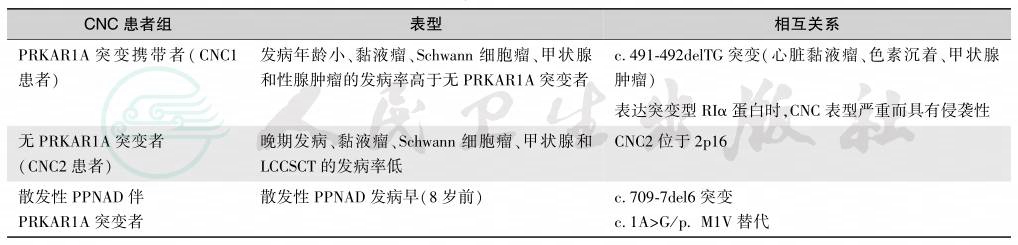
注:CNC:Carney complex,Carney复合症;PPNAD:primary pigmented nodular adrenocortical disease,原发性色素性结节性肾上腺皮质病;PRKAR1A:protein kinase A regulatory subunit 1α,蛋白激酶A调节亚基1α
PRKAR1A的功能丢失可引起PKA途径过度活跃。在垂体中,GHRH受体通过cAMP/PKA途径刺激GH的合成与分泌。GNAS活化性突变导致GH瘤。
CNC的诊断标准见表4。
表4 Carney复合症的诊断标准

注:CNC:Carney complex,Carney复合症;PPNAD:primary pigmented nodular adrenocortical disease,原发性色素性结节性肾上腺皮质病;LCCSCT:large-cell calcifying Sertoli cell tumor,大细胞钙化性Sertoli细胞瘤;PRKAR1A:protein kinase A regulatory subunit 1α,蛋白激酶 A 调节亚基1α
(一)病例筛查
一般出现下列情况应进行Carney复合症的排查:①皮肤黏膜上加深的斑点,但无典型分布或变黑的色素斑;②多发性普通型蓝痣;③咖啡斑或棕黄斑或其他“胎记”;④血IGF-1升高或OGTT异常或临床上无肢端肥大症的表现,但TRH兴奋试验GH反应失常;⑤心肌病;⑥多毛窦;⑦Cushing综合征、肢端肥大症和猝死的家族史;⑧癌肿(特别是甲状腺、大肠、胰腺和卵巢癌)家族史;⑨年轻患者有单个良性甲状腺结节,年老患者有多发甲状腺结节;⑩其他多发的良性或恶性肿瘤和症状轻微或无症状的高PRL血症等。结合临床和分子生物学的检测可达到早发现、早诊断和早治疗的目的。2001年,Stratakis等总结了Carney复合症从发现以来所报道的病例,并对其症状、体征及家族史进行汇总分析,制订出主要和次要的诊断标准,患者如具有主要标准的任意2项或具有1项主要标准和1项次要标准即可临床诊断Carney复合症。
1.肾上腺皮质功能测定
因原发性色素性结节性肾上腺皮质病相对常见,应重点或优先测定肾上腺皮质功能。血皮质醇明显升高、节律紊乱,ACTH不能测出,24小时尿游离皮质醇也明显升高和8mg地塞米松抑制试验不能抑制,提示PPNAD的存在。睾丸或卵巢受累时,也可出现血E2和睾酮降低,而LH和FSH可升高。
2.垂体检查
不论有无垂体瘤表现,均应排除GH瘤和PRL瘤可能。TRH刺激生长激素分泌试验中,GH对TRH刺激有反应,提示可能存在垂体GH瘤。在有GH瘤的CNC患者中,血IGF-1和GH升高。血PRL升高,提示存在高PRL血症或PRL瘤。OGTT可异常。
3.病理检查
肾上腺皮质黑褐色结节和巨大脂质球状皮质细胞是较特异的病理发现。手术后肿瘤病理学检查示单侧或双侧肾上腺皮质呈现黑褐色的小结节状增生。显微镜下,结节主要由巨核、胞质嗜酸性和含有脂质的巨大球状皮质细胞组成对PPNAD有确诊意义。病理学检查对黏液瘤也有确诊价值。镜下,黏液瘤主要由间质细胞、黏液性基质成分、胶原、肥大细胞和炎症细胞组成。
4.影像检查
B超可发现黏液瘤、卵巢囊肿或肿瘤、乳腺肿瘤、睾丸肿瘤和甲状腺结节或肿瘤。垂体可有或无肿瘤征象。腹部CT或MRI可发现单侧或双侧肾上腺多结节性增生。
(二)鉴别诊断
1.咖啡斑
咖啡斑见于McCune-Albright综合征、多发性神经纤维瘤病、Carney复合症、Peutz-Jeghers综合征和正常人。Carney复合症患者有色素痣和斑点状色素沉着,类似于McCune-Albright综合征和von Recklinghausen病(多发性神经纤维瘤病)的棕黄斑或咖啡斑(café-au-lait spot),但Carney复合症的皮肤颜色变浅、面积小而且趋向于面部中央密集,通常随增龄而消退。同时还要与普通的雀斑、“胎记”或痣鉴别,后几者无黏液瘤或多发性内分泌腺肿瘤的证据,可资鉴别。Peutz-Jeghers综合征多见于儿童和青少年,是一种常染色体显性遗传病,患者有胃肠道多发性错构瘤样息肉,手足皮肤及口腔黏膜的色素沉着,其特点是嘴唇与口腔黏膜出现边界清晰的黑色素痣(lentigene)或色素斑,故又称为皮肤黏膜黑斑息肉病。有些男性患者还伴有睾丸钙化及乳腺发育。肠息肉最常见于小肠,不具备Carney复合症的主要特点(黏液瘤和其他内分泌肿瘤),可资鉴别。
2.多发性内分泌腺肿瘤
多发性内分泌腺肿瘤可见于MEN、多发性神经纤维瘤病、von Hippel-Lindau病和Carney复合症。Carney复合症患者可有原发性色素沉着性结节性肾上腺皮质病(PPNAD)、大细胞钙化性支持细胞瘤(LCCSCT)、垂体GH/PRL瘤、甲状腺腺瘤、卵巢肿瘤或卵巢囊肿,需要与上述疾病鉴别,上述疾病不具备诊断Carney复合症的其他主要诊断标准和次要诊断标准(表5)。von Hippel-Lindau病的特点是家族性视网膜或中枢神经系统成血管细胞瘤,嗜铬细胞瘤合并肾脏、胰腺、附睾等部位的肿瘤。微结节肾上腺病可能伴或不伴CNC,其临床鉴别见表6。
表5 Carney复合症的诊断依据

注:PPNAD:原发性色素性结节性肾上腺皮质病;LCCSCT:大细胞钙化性Sertoli细胞肿瘤;PRKAR1A:区域内cAMP依赖性蛋白激酶A α调节亚基。Liddle试验:标准的Liddle试验是将大剂量、小剂量地塞米松抑制试验合并使用。即先每6小时口服地塞米松0.5mg,连续2天,接着每6小时口服地塞米松2mg连续2天,测定用药前和用药后尿游离皮质醇和17-羟类固醇。正常者尿游离皮质醇下降超过90%,而17-羟类固醇下降超过60%
表6 微结节肾上腺病伴或不伴Carney复合症的特点

注:微结节性肾上腺皮质病的结节为多发性,结节直径<1cm,可能伴有或不伴有CNC;CNC:Carney complex,Carney复合症;iPPNAD:isolated primary pigmented nodular adrenocortical disease,单纯性原发性色素结节性肾上腺皮质病;C-PPNAD:primary pigmented nodular adrenocortical disease associated with Carney complex,原发性色素结节性肾上腺皮质病伴Carney复合症;MAD:adrenocortical disease,肾上腺皮质病;PDE:phosphodiesterase,膦酸二酯酶;PRKAR1A:protein kinase A regulatory subunit 1α,蛋白激酶A调节亚基1α
3.着色斑病综合征的病因鉴别
见表7。着色斑病综合征的发病机制与细胞的下游信号途径PKA、Ras-MAP激酶和mTOR相关。这些信号分子突变后,因生长、增殖、分化异常而导致肿瘤。着色斑(亦称斑痣,lentigine),是指皮肤和黏膜上的平坦性色素性斑点,通常的直径<0.5cm,边缘不规则,不连续,色泽深浅不一,通常为棕色或黑色。家族性着色斑病综合征患者的神经-内分泌系统与间质常发生肿瘤。组织学检查可见上皮显著增厚,皮肤基底细胞色素沉着,黑色素细胞增生。而雀斑(freckles)的组织学特点是黑色素细胞数目正常,色素沉着仅因为皮肤的基底角质细胞黑色素增多,发病部位仅限于阳光暴露部位。着色斑可发生于身体的任何部位,如结膜、嘴唇、大阴唇、手掌、足底等处;接触阳光后,色素不进一步加深,其中以Peutz-Jeghers综合征最常见,但亦可见于其他遗传综合征,如Carney复合症、Laugier-Hunziker综合征、Ruvalcaba-Myhre-Smith综合征、Bannayan-Zonnana综合征、Cowden病或LEOPARD/Noonan综合征(图9)。
表7 伴有家族性着色斑病的遗传综合征
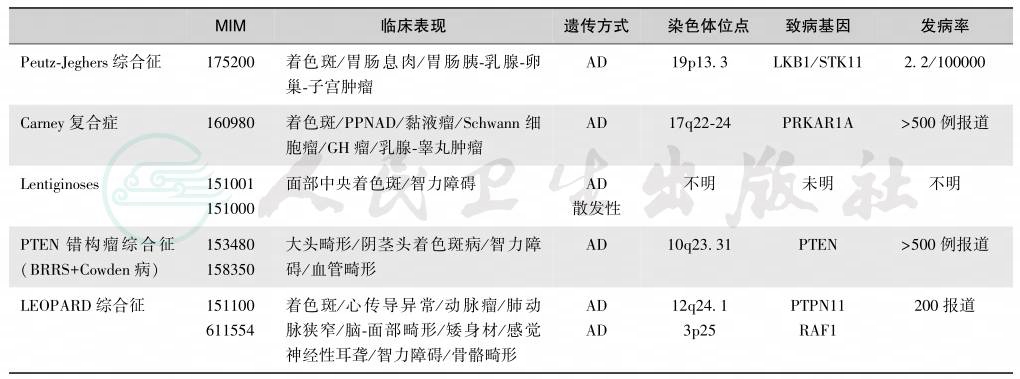
注:lentiginoses:着色斑病;lentigines:着色斑;PPNAD:primary pigmentednodular adrenocortical disease,原发性色素结节性肾上腺皮质病
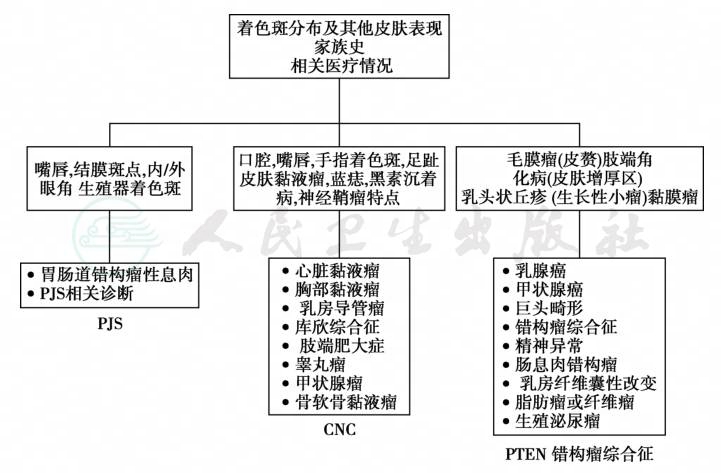
图9 着色斑/着色斑病的鉴别诊断
重点在于治疗内脏肿瘤。心脏黏液瘤应早期手术治疗,经膈途径切除左心房黏液瘤可预防复发;复发者可再次手术。PPNAD首选治疗为双侧肾上腺切除术,术后用泼尼松或氟氢可的松替代治疗,若手术有禁忌证而不能手术者,可选用美替拉酮、氨鲁米特或酮康唑等治疗。睾丸肿瘤若无转移则以保守治疗为宜。垂体瘤可手术治疗。









