


 收藏
收藏 已收藏
已收藏英文名称 :multiple endocrine adenoma-Ⅱ syndrome
中文别名 :西普勒综合征
1959年,Sipple首先报告2型多发性内分泌腺肿瘤综合征(MEN-2),故又称为Sipple综合征。
(一)根据临床表型分为MEN-2A和MEN-2B两种
过去曾将MEN-2B称为MEN-3型,但从病因看,MEN-3应属于MEN-2中的一种亚型。在MEN-2A中还有两种变异型,各型名称及临床表型见表1。
表1MEN-2表型
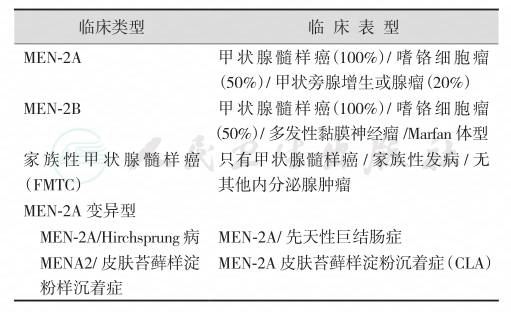
有些学者把FMTC作为MEN-2中的一种亚型(根据病因)或变异型(根据表型)。
(二)RET突变导致MEN-2
根据对MEN-2多个家族进行连锁分析,1993年已确定MEN-2综合征的突变基因在10号常染色体的长臂上,即10q11.2的RET原癌基因(RET proto-oncogene),其表达产物为RET蛋白。RET原癌基因主要在胚胎神经嵴的神经内分泌细胞和神经母细胞肿瘤中表达,对神经细胞增殖、分化和肾脏的生成起作用。
RET原癌基因长55kb,由21个外显子和18个内含子组成。外显子1~10负责编码RET蛋白的细胞外区,其余的外显子负责编码RET蛋白的细胞内区和尾部。RET蛋白存在于细胞膜表面,表达于由胚胎神经嵴衍化而来的细胞属于酪氨酸激酶家族中的成员。没有糖基化的RET蛋白分子量120kD,糖基化RET为170kD。RET蛋白的结构已研究清楚,像其他酪氨酸激酶一样,RET在细胞外区有一类黏附蛋白区(cadherin),接着为一富含半胱氨酸的细胞外区,细胞内区为酪氨激酶1区和2区。在富含半胱氨酸的细胞外区有5个半胱氨酸,其中4个由外显子10编码,它们的位点分别为609、611、618和628。另一个半胱氨酸由外显子11编码,位点是634。98%的MEN-2的发病都是由于RET原癌基因胚系(germ-line)突变。MEN-2A中最常见的突变为534位的半胱氨酸被其他氨基酸取代,在FIMTC中以634的突变居多(占79%),其他少见的部位为外显子13的768位、外显子14的804位和630位以及外显子15的883位突变。在MEN-2B中则只有外显子16的918位点突变。所有引起MEN-2综合征的RET原癌基因突变均为单碱基突变。
在先天性巨结肠(Hirschsprung病)中,有外显子10的TGC609TAC及TGC620CGC两种突变,使RET蛋白中的半胱氨酸分别被酪氨酸和精氨酸取代;而在MEN-2A/Hirschsprung病表型中则有密码子10、609位点有与Hirschsprung病相同的突变。另外,外显子10的TGC618AGC突变使RET蛋白在相应位置的半胱氨酸被丝氨酸取代。在国际RET突变合作分析中,477个MEN-2家族中有6个家族为MEN-2A/ Hirschsprung病,其中在620(5个家族)和618位点(1个家族)有突变,RET蛋白相应位置上原有半胱氨酸被精氨酸取代,有18个家族中为MEN-2A/CLA,且CLA可发生于MEN-2A确诊前。在MEN-2B中未发现CLA患者。
国际RET突变合作研究发现,186个家族中160例患有MTC和嗜铬细胞瘤,均有634位点突变,而43例未患嗜铬细胞瘤的家族中只有18个家族有634位点突变,提示密码子634突变与家族中嗜铬细胞瘤相关。密码子634半胱氨酸被精氨酸取代(TGC→CGC),则与甲旁亢联系。
(三)RET突变导致受体复合物二聚化和酪氨酸激酶激活
胶质细胞衍生的神经营养因子(GDNF)是转化生长因子大家族成员,其受体为GDNF-α。在没有配体存在情况下,RET受体和GDNF-α受体以非二聚体存在,在有配体存在时则GDNF与GDNF-α受体结合,但GDNF也可与RET受体相互作用,使RET二聚体化而引起自身磷酸化和酪氨酸激酶激活。在634密码子突变中,RET中的半胱氨酸被取代出来而无“搭档”,这种无“搭档”的半胱氨酸很易与另一个具有相同突变的ret相互结合,从而使ret二聚体化而导致酪氨酸激酶的激活和自动磷酸化。
RET突变可分为富含半胱氨酸的胞外结构域(cysteinerich extracellular domains)突变和非半胱氨酸的胞外结构域(non-cysteine intracellular domains)突变两种类别,一般前者的临床表现较为典型,而后者的表现更隐匿,但基因型与表型的关系复杂,不均一性明显。除menin蛋白外,还有许多决定临床表型的因素,这些因素统称为MEN-1相关性组织选择性肿瘤形成决定因子(factors determining NEN-1-associated tissue-selective tumorigenesis),如混合型白血病蛋白-含组蛋白的甲基转移酶(mixed-lineage leukemia protein-containing histone methyl transferase,MLL-HMT)。
有些突变可直接使酪氨酸激酶激活,如外显子16的918位和883位突变。由此看来,MEN-2各种表型的发病机制可能是不同的。
(一)五肽胃泌素和RET突变分析筛查家族成员
对MEN-2A家族成员必须从1~35岁每年作1次试验,但可出现假阳性和假阴性结果。除此之外,还要每年测定24小时尿肾上腺素和去甲肾上腺素以筛查有无嗜铬细胞瘤;每2年要测血清钙或离子钙以筛查MEN-2A家族成员和突变基因携带者有无甲状旁腺功能亢进症。在诸多筛查中以筛查RET基因突变为最可靠的方法。
对MEN-2B患者的家族成员应进行RET基因突变筛查,对突变基因携带者应尽早做甲状腺全切术以预防以后发生甲状腺髓样癌,并终生追踪观察。分子遗传学检查不仅有助于诊断,而且可以精确地检出突变基因的携带者。
(二)手术切除内分泌腺肿瘤
突变基因携带者尽管目前无临床表现,也必须作甲状腺预防性切除。MEN-2A应在5岁前,MEN-2B则在1岁时作此种手术,不过不能单独根据五肽胃泌素试验阳性决定,而应重复作分子遗传学分析以确诊为突变基因的携带者。手术时除将甲状腺全部切除外,靠近甲状腺的淋巴结也要全部切除,颈部两侧的淋巴结则对可疑有病者作选择性切除。术后应终生用甲状腺激素替代治疗。且对手术患者应追踪有无复发。可定期作五肽胃泌素试验,阴性结果表明治愈,阳性结果则表明有复发。复发的来源有甲状腺部位和转移灶,术前应作出定位。定位的方法有放射性铊和MIBG扫描、奥曲肽(octreotide)扫描和静脉插管在不同解剖部位采血测CT。
因为MEN-2中嗜铬细胞恶变者少,故如果只检出一侧肾上腺有嗜铬细胞瘤,则只切除一侧肾上腺,另一侧定期随访,出现肿瘤后再作手术切除。一侧肾上腺嗜铬细胞瘤手术者除经腰部途径外,亦可采用腹腔镜切除肿瘤。术前应给予α和β肾上腺素能拮抗剂准备。双侧肾上腺切除者应终生补充生理剂量的糖皮质激素,遇应激时应适当增加剂量。
如果患者同时有甲状腺髓样癌和嗜铬细胞瘤,则应先作嗜铬细胞瘤手术,后作甲状腺髓样癌手术,以免作后种手术时在术中诱发高血压危象或休克。
(三)根据病情选择巨结肠治疗方法
内科治疗可采取扩肛、服缓泻剂,辅以灌肠,适用于轻度患者。也可采用结肠灌洗,即用一肛管从肛门插到扩张的肠管处,用等渗盐水来回冲洗,每天2~3次。手术方法有结肠造瘘和部分直肠、结肠切除再作直肠结肠吻合。此病易并发小肠结肠炎,死亡率高。









