


 收藏
收藏 已收藏
已收藏15%~20%的家族型嗜铬细胞瘤是由于RET突变所致。RET为原癌基因,其活化往往有利于肿瘤的发生。MEN-2A患者表现为肾上腺嗜铬细胞瘤、甲状腺髓样癌及甲状旁腺腺瘤。MEN-2B患者除肾上腺嗜铬细胞瘤(双侧性)、甲状腺髓样癌及甲旁亢外,还有黏膜神经瘤、角膜神经增厚、肠神经节神经瘤病(intestinal ganglioneuromatosis)和Marfan综合征样外形。儿童嗜铬细胞瘤主要位于肾上腺外组织,且多为家族性、双侧性与多灶性。
MEN-1为肿瘤抑制基因MEN-1失活性突变所致,呈常染色体显性遗传,其临床特点是垂体瘤、原发性甲旁亢、胰岛细胞瘤、血管纤维瘤和脂肪瘤,偶尔合并嗜铬细胞瘤。
VHL是一种抑癌基因,编码两种蛋白。一种含213个氨基酸,分子量28~30kD(pVHL30);另一种含160个氨基酸。分子量18kD(pVHL19)。目前已发现200多种突变类型,突变基因所表达的蛋白有丧失功能和获得功能两种。70%~90%的患者为种系突变,其他为体细胞突变;前者决定VHL家族的肿瘤易感素质及发病情况,而后者与肿瘤的恶性倾向有关。von Hippel-Lindau病患者可发生嗜铬细胞瘤(10%~90%),常为多发性。
Carney复合症常发于青少年,发病与Carney复合症基因突变有关,可伴有间叶细胞瘤(尤其是心房黏液瘤)、皮肤色素沉着和外周神经损害。
线粒体琥珀酸脱氢酶(succinate dehydrogenase,SDH)是三羧酸循环和有氧电子传递呼吸链中的关键酶之一,包含A、B、C、D等4个亚基。在家族性和散发性嗜铬细胞瘤中存在SDHD、SDHB基因突变。
嗜铬细胞瘤虽然罕见,但是血压和心功能急性改变的重要原因。这些肿瘤通常是单侧的,但是可能发生在两侧和肾上腺髓质外,如交感神经节的任何地方。从肿瘤中释放去甲肾上腺素,肾上腺素导致心血管的严重症状和体征。
嗜铬细胞瘤亦称肾上腺髓质副神经节瘤,肾上腺外的嗜铬细胞瘤亦称嗜铬性副神经节瘤。嗜铬细胞瘤常为单侧单发性,偶为双侧性或多发性,或肾上腺内‐肾上腺外多发性。肿块大小不一,直径5~6cm,圆形,境界清楚。典型病例切面灰棕色,可有出血、囊性变和钙化(图1)。新鲜肿瘤组织经重铬酸钾处理后呈棕色。瘤细胞与正常的嗜铬细胞相似,胞质嗜铬性,含PAS阳性物,但常见巨核和核浓染,这并非恶性征象。电镜见瘤细胞胞质中含许多致密核心的神经分泌颗粒。免疫组化分析示细胞有肾上腺素、去甲肾上腺素、多巴胺、儿茶酚胺合成酶、NSE、铬粒素和突触素等的阳性表达。嗜铬细胞瘤还可表达三种不同分子量的神经纤维微丝蛋白(NP)。瘤细胞周围的支持细胞呈S‐100蛋白强阳性反应。
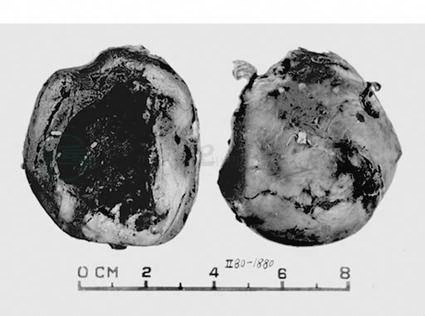
图1 肾上腺嗜铬细胞瘤
确诊是通过直接测量血尿儿茶酚胺、肾上腺素和去甲肾上腺素的代谢产物,其中包括3-甲氧基肾上腺素。









