


 收藏
收藏 已收藏
已收藏英文名称 :aldosterone insensitivity syndrome
中文别名 :假性低醛固酮血症
醛固酮不敏感综合征(aldosterone insensitivity syndrome)又称假性低醛固酮血症(pseudohypoaldosteronism,PHA),是一种血清电解质代谢紊乱的异质性疾病,以肾小管对醛固酮作用无反应(抵抗)、高血钾、代谢性酸中毒和肾小球滤过率正常为特征。临床上存在不同的醛固酮不敏感综合征类型,患者可出现血容量不足或过多,失盐或盐潴留,血压降低或升高,醛固酮正常、升高或降低等多种不同的临床表现。
(一)钠通道亚基突变
上皮细胞钠通道(ENaC)亚基的活化性突变导致高血压,并在妊娠期明显加重,这事实上是一种盐皮质激素过敏感综合征。而β和γ亚基的作用是促进α亚基的表达。ENaC的3个亚基对钠的转运均是必需的,只有当ENaC三个亚基同时表达才可获得最大的钠通透性。ENaC分子含两个高度保守的富含半胱氨酸的细胞外袢,即CRD1和CRD2区,它们是维持Na+转运功能的最关键结构。β或γ亚基基因的活化性突变(功能亢进)引起Liddle综合征。相反,Ⅰ型常染色体隐性遗传性假性低醛固酮血症是由于ENaC的α、β或γ亚基的失活性突变(功能减弱)所致。此外,肺水肿和囊性纤维化也与ENaC的调节障碍有关。
1.Ⅰ型醛固酮不敏感综合征
又可分为肾型醛固酮不敏感综合征-Ⅰ和多靶器官缺陷型醛固酮不敏感综合征-Ⅰ(multiple target organ defect,MTOD醛固酮不敏感综合征-Ⅰ)。Ⅰ型醛固酮不敏感综合征临床表现多样,幼儿期高钾血症又称为Ⅳ型肾小管酸中毒第5亚型,属于肾型醛固酮不敏感综合征-Ⅰ的一个亚型。Ⅰ型醛固酮不敏感综合征为一种遗传性疾病,遗传方式为常染色体显性或隐性遗传。常染色体显性遗传者仅有肾小管对醛固酮作用抵抗表现,而隐性遗传者更严重,且累及汗腺、唾液腺及结肠等,钠不能转运入细胞内以致从大便、小便、汗液和唾液中丢失而导致低钠血症。低钠血症加上血容量减少,使RAA被激活,故有血浆醛固酮和肾素升高,由于肾小管不能重吸收钠,以致在肾远曲小管中的钠-钾交换减少,故有高钾血症。真性低醛固酮症或表观低醛固酮症(apparent hypoaldosteronism)主要表现为肾小管性酸中毒伴高钾血症,多见于糖尿病患者,亦可见于肾素/醛固酮生长不足的其他许多疾病,如前肾素原突变(如p.Cys20Arg)。
2.Ⅱ型醛固酮不敏感综合征
又称Gordon综合征。其缺陷不在醛固酮受体,而是远端肾小管对氯的重吸收增多,从而抑制了钠及醛固酮介导的钾-氢分泌,导致高钾血症和高氯性酸中毒。因为氯化钠的重吸收增多,引起血容量增多和血压升高,使肾素-醛固酮系统受到抑制。
(二)肾病和药物
继发性醛固酮不敏感综合征又称为暂时性或可逆性醛固酮不敏感综合征,通常是由于某些肾脏疾病和药物所引起。主要见于一些泌尿生殖系统疾病,如阻塞性尿路疾病、急性肾盂肾炎、尿路感染、小管间质性肾病、系统性红斑狼疮、肾静脉血栓形成或肾髓质坏死等。多发性骨髓瘤或肾移植术后的患者也可能出现继发性醛固酮不敏感综合征。发生醛固酮不敏感综合征的机制未明,可能与上述疾病造成肾小管损害,而导致肾小管对醛固酮的反应性降低有关。随着原发病的治愈,一般醛固酮的敏感性可逐渐恢复正常。
尿道畸形合并感染时,可并发一过性1型假性低醛固酮症(transient type 1 pseudo-hypoaldosteronism,TPHA1),本症主要发生于婴幼儿,表现为低钠血症、高尿钠症、低尿钾症、高钾血症、代谢性酸中毒和血肌酐升高;血清醛固酮升高提示肾小管对醛固酮存在抵抗。
(三)MRENaC突变
醛固酮敏感的靶细胞包括肾远曲小管、汗腺、结肠黏膜上皮、唾液腺及泪腺细胞。除经典的核受体作用外,醛固酮还表现出许多非核受体作用,并与心钠素-肾素-醛固酮系统偶联,如调节靶细胞的离子转运,参与心肌细胞的代谢和重建等。虽然MR与醛固酮和皮质醇结合的亲和力相等,但在这些醛固酮的靶组织中,由于存在Ⅱ型 11β-羟类固醇脱氢酶(11β-HSD2,CYP11B2),可以使皮质醇转化成没有活性的皮质素,从而使MR只能被醛固酮所活化。但在表达MR的组织中,如果缺乏11β-HSD2(如中枢神经的神经细胞),MR仍主要由皮质醇激活。Ⅰ型醛固酮不敏感综合征中的肾型主要是由于MR基因突变而导致MR的数量或功能缺陷所引起;Ⅰ型醛固酮不敏感综合征中的多靶器官缺陷型主要是由于ENaC的α或β亚基的失活性突变(功能减弱或丧失),导致存在ENaC的组织器官(肾、肺、结肠、汗腺、唾液腺)的钠离子转运缺陷。故Ⅰ型醛固酮不敏感综合征又可分为肾性和多靶器官缺陷型性。Ⅰ型醛固酮不敏感综合征有两种遗传方式。一类为常染色体隐性遗传或散发,患者自幼发病并持续到成年期,病变在腔侧面的阿米洛利(amiloride)敏感性钠通道(MTOD PHA-Ⅰ);另一类为常染色体显性遗传或呈散发性,症状轻,至成年后可完全或部分缓解,其中一些患者是由于MR基因突变所致(肾型醛固酮不敏感综合征-Ⅰ),见表1。
表1 盐皮质激素受体突变所致的Ⅰ型醛固酮不敏感综合征
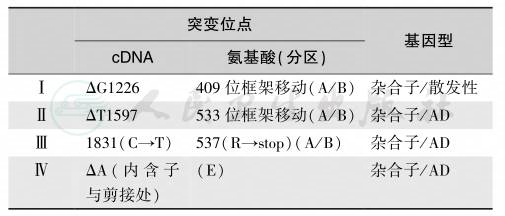
注:氨基酸突变位点的分区是指MR分子的A~E分区,即A/B区为免疫活性区,C区为DNA结合区,D区和E区为配体结合区。AD:常染色体显性遗传
钠通道γ亚基1基因位于人类染色体16p12,编码阿米洛利敏感性上皮细胞钠通道-1蛋白质,该基因的突变引起Liddle综合征(活化性突变)或Ⅰ型醛固酮不敏感综合征(失活性突变为1627缺失G,导致外显子后提前终止编码和1570G→A,两侧均为复合性杂合子)。
(四)远端肾小管氯重吸收增多
Ⅱ型醛固酮不敏感综合征的缺陷抑制了钠离子及醛固酮介导的钾、氢离子的分泌,因而导致高钾血症和高氯性酸中毒。目前认为Ⅱ型醛固酮不敏感综合征的遗传方式为常染色体显性遗传,也有散发病例。Mansfield TA等通过对8个发病家系的研究,将醛固酮不敏感综合征-Ⅱ的致病基因定位于1q31-q42和17p11-q21。后来发现,12p13.3也与醛固酮不敏感综合征-Ⅱ发病相关。新近发现亦与WNK1激酶及WNK4激酶基因突变有关。WNK激酶存在于多细胞生物体中,与其他蛋白激酶相比,它缺乏1个关键的赖氨酸残基。WNK激酶对肾脏的钠、钾转运起调节作用。WNK激酶-1(位于细胞质中)的突变,是由于内含子的缺失突变,使WNK1表达增加5倍。WNK4(位于紧密连接处)是错义突变,可以引起NaCl联合转运体(NCCT)过度表达以及内向性钾通道调校蛋白(ROMK)表达缺乏,从而导致盐潴留、高血压和高钾血症。
(五)肾小管对醛固酮反应性丧失
钠和水从大小便大量丢失而导致低钠血症和血容量减少,继而使醛固酮代偿性分泌增多,继发性醛固酮不敏感综合征。肾脏和生殖器疾病可引起继发性醛固酮不敏感综合征,可能是由于肾小管损害使肾小管对醛固酮反应性丧失,钠和水从大小便大量丢失而导致低钠血症和血容量减少,继而使醛固酮代偿性分泌增多,以减少尿钠排泄和增加结肠中钠和水的吸收,但这种代偿不足以使血清电解质得以纠正,从而表现出对醛固酮作用的抵抗。婴幼儿的继发性醛固酮不敏感综合征是由于肾小管尚未发育成熟,对醛固酮的反应性本身就差,当某些疾病累及肾小管时,加剧了这种情况,而诱发醛固酮不敏感综合征。
(1)电解质:
血清钠降低,血钾升高,但是如果患者有明显脱水,血液浓缩,低钠可以被掩盖。尿中钠钾排泄与血清相反,钠增多,钾减少。如果结肠、汗腺和唾液腺也受累,则大便、汗腺和唾液中也有与尿中相同的钠钾变化,使钠钾比值增大。有时候会出现尿钙过多。Ⅱ型患者有血清氯化物增高,Ⅱ型和继发性者可有CO2CP降低。肾功能及肾小球滤过率正常。
(2)肾素和醛固酮:
Ⅰ型患者血浆PRA可高达6500pg/ml(正常50~800pg/ml),每日摄入正常或低于正常钠时和服用螺内酯均可使肾素和醛固酮进一步升高。血浆去氧皮质酮、皮质醇浓度正常,18-羟皮质酮与醛固酮的比值正常,尿醛固酮及其代谢产物如四氢醛固酮增高。Ⅱ型患者PRA和醛固酮降低或正常,如果给予利尿剂或限盐后,高血容量被纠正,血PRA和醛固酮升高。
(3)X线及超声检查:
在MTOD醛固酮不敏感综合征-Ⅰ的患者,胸部X线可有呼吸道液体过多的表现,类似于囊性纤维病。Ⅰ型患者可有肾钙质沉着,Ⅱ型患者可有肾石病。
(4)MR功能及基因突变分析:
有少数报告患儿结肠黏膜上皮和周围血单核细胞的MR数目和(或)亲和力降低。但MR数目和(或)亲和力降低的原因(即MR基因突变)为何,目前尚缺乏研究。必要时应进行MR基因的突变分析,阐明本综合征的分子病因。
Ⅰ型醛固酮不敏感综合征的治疗主要是补钠和处理高钾血症。一般每天补钠8~50mmol/kg,才可达到钠平衡。随着年龄增长,每天补钠量则可逐渐减少;大多数患儿到2岁时可停止补钠而无症状,尽管此时血浆PRA和醛固酮浓度仍保持在较高水平。一般在饮食中增加氯化钠量,但在病情危急时则应静脉补充0.9%生理盐水或重碳酸氢钠溶液。MTOD醛固酮不敏感综合征-Ⅰ型患者还应该给予低钾饮食(每天 0.6mmol/kg)。
血钾过高常引起威胁患者生命的严重心律不齐或停搏,此时应采取紧急措施进行抢救。除静脉补以钠盐外,还可口服磺酸乙烯苯树脂作肠道透析将钾从大便排出;亦可采用血液透析或在静脉滴注的10%葡萄糖生理盐水中加入适量胰岛素,促使血钾转入细胞内。可在抢救危重患者时短期应用前列腺素抑制剂,并注意纠正酸中毒。
Ⅱ型醛固酮不敏感综合征的治疗与Ⅰ型醛固酮不敏感综合征基本相同,但纠正酸中毒后不能改善高血钾。患者酸中毒和高钾血症的处理与Ⅰ型醛固酮不敏感综合征相同,但此型患者纠正酸中毒后并不能纠正高血钾,因而限钠的同时要限制钾的摄入,氢氯噻嗪或呋塞米(速尿)利尿后可降低血钾、改善酸中毒并降低血压。









