


 收藏
收藏 已收藏
已收藏英文名称 :17α-hydroxylase deficiency
中文别名 :17α-羟化酶缺陷症
17α-羟化酶乏症即 17α-羟化酶 /17,20-裂解酶缺乏症(17-OHD,OMIM:202110)是一种罕见的先天性肾上腺皮质增生症(CAH)类型,是由CYP17A1基因缺陷引起的一种常染色体隐性遗传病。CYP17A1基因编码17α-羟化酶和17,20-裂解酶。17α-羟化酶缺陷导致皮质醇生成减少,引起促肾上腺皮质激素分泌增加,从而导致11-脱氧类固醇激素的生成增加。由于17,20-裂解酶活性受损导致雄激素生成减少,使雄激素和雌激素联合缺乏。17-OHD通常分为2种临床类型:完全型和部分型。完全型临床主要表现为高血压、低血钾、第二性征发育不良,其中46,XX患者呈性幼稚、原发性闭经,46,XY患者呈女性外生殖器或假两性畸形。
17-OHD占所有CAH病例的不到1%。然而,根据Fontenele对近80例已知病例的调查,17-OHD是巴西人群第二常见的CAH,占所有CAH病例的近7%。
细胞色素P450c17酶兼具羟化酶(17α-羟化酶)和裂解酶(17,20-碳链裂解酶)2种活性。17α-羟化酶负责将孕烯醇酮和孕酮17α-羟化,分别转化为17-羟孕烯醇酮和17-羟孕酮,而后在不同酶的作用下合成皮质醇和性激素。17,20-碳链裂解酶将17-羟孕烯醇酮和17-羟孕酮的17,20-碳链裂解,转化为DHEA及雄烯二酮,进而合成睾酮、雌二醇等性激素。17α-羟化酶活性缺乏时,孕烯醇酮和孕酮不能转化为17-羟孕烯醇酮和17-羟孕酮,皮质醇合成受阻,ACTH反馈性增加,刺激双侧肾上腺皮质增生。同时,在ACTH的刺激下,以及由于前体物质孕烯醇酮和孕酮的堆积,盐皮质激素通路中的11-脱氧皮质酮(DOC)和皮质酮大量增加。DOC可比参考范围高1 000倍,皮质酮可增高50~100倍。DOC有较强的理盐作用,作用于盐皮质激素受体,使肾脏集合管保留钠离子,导致水钠潴留、血容量增加,抑制肾素-血管紧张素系统,导致低肾素性高血压、醛固酮水平低下;同时DOC使肾脏集合管钾离子排出增加,导致低钾血症。此外,皮质酮也具有弱糖皮质激素活性,因此尽管皮质醇水平很低,患者一般不会表现出肾上腺功能不全的症状。
17,20-碳链裂解酶活性缺乏时,17-羟孕烯醇酮和17-羟孕酮不能转化为DHEA及雄烯二酮,性激素合成受阻,促性腺激素LH和FSH反馈性增高。通常男性患者具有Y染色体及SRY基因,能形成睾丸。睾酮由睾丸Leydig细胞生成,其中起重要作用的酶是细胞色素P450c17酶,P450c17酶缺陷使雄激素分泌不足,导致睾丸发育停滞,不能形成阴茎和阴囊,外生殖器保持为幼女型。睾丸Sertoli细胞产生米勒管(Müllerian duct)抑制因子,米勒管正常退化,无子宫、输卵管和阴道上段的发育,阴道呈盲端。女性患者不能形成睾丸,而有子宫及其附件的结构,外阴呈女性,但因青春期雌激素合成不足,导致第二性征发育不良,常无月经来潮。此外,因性激素合成不足,患者骨骺闭合延迟,骨龄小于实际年龄,且常伴有骨质疏松。
CYP17A1基因是细胞色素P450超基因家族17亚族(P450c17)的唯一成员,位于染色体10q24.3-q25上。CYP17A1基因在人体肾上腺和性腺中都有表达,因此17-OHD既损伤肾上腺功能又损伤性腺功能。CYP17A1基因全长约6.6kb,包括8个外显子和7个内含子。该基因在肾上腺和性腺中均转录的mRNA,长度为2.1kb,其编码区长1.6kb,编码由508个氨基酸残基组成的蛋白酶,即细胞色素P450c17酶。影响CYP17A1基因的不同突变,其中大多数(62%)是错义/无义突变。在巴西人群中,17-OHD患者(70个等位基因)中,p.W406R(40%)和p.R362C(28%)是两种最常见的突变。
(一)实验室检查
皮质醇、性激素和17-羟孕酮水平降低,孕酮、DOC和皮质酮水平升高。由于皮质醇和性激素缺乏,LH、FSH和ACTH水平明显升高。由于DOC具有盐皮质激素的作用,肾素水平受抑制,醛固酮和血钾降低。
(二)基因检测
CYP17A1基因突变对明确诊断至关重要。迄今为止,已在人类基因突变数据库中发现140多个CYP17A1突变,包括错义和无义突变、插入、缺失和剪接位点变异,尚无明确的热点突变。但某些突变高频出现在特定人群(表1):①在荷兰弗里斯兰人的后代中发现了导致移码的4个核苷酸的重复;②在东南亚发现了残基487~489的框内缺失;③在53或54位发现了苯丙氨酸的缺失;④在西班牙和葡萄牙血统的巴西人中发现W406R和R362C突变。CYP17A1基因型和临床型之间的关系尚不清楚。
表1 17-OHD中CYP17A1基因常见突变位点
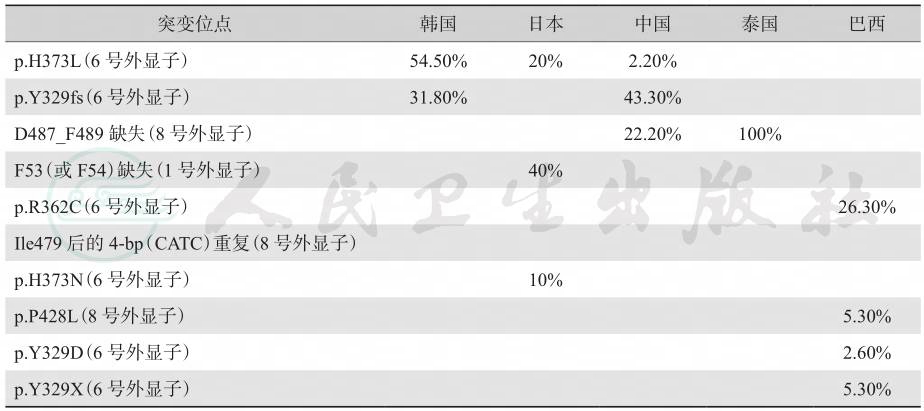
注:表中空格表示无相关数据。
(三)影像学检查
盆腔彩超,女性患者往往提示幼稚子宫和反复发生的卵巢囊肿;外表为女性的46,XY男性患者不能探及子宫、卵巢,但腹腔或腹股沟内可有发育不良的睾丸。CT检查可见双侧肾上腺增生或正常。X线片往往提示骨龄延迟。
经典型17-OHD的治疗目标是减轻盐皮质激素过度分泌的影响、预防糖皮质激素缺乏,以及恢复第二性征并获得相应的益处,比如改善骨密度(bone mineral density,BMD)。除了超生理剂量的糖皮质激素以外,还可以通过阻断盐皮质激素及补充生理剂量的皮质醇和性激素来实现。
大多数患者规律使用糖皮质激素治疗后,血钾、血压恢复正常;少部分患者需加用降压药物达到血压控制目标,这可能与长时间高血压已经造成心血管损害有关。治疗较晚或治疗不规律的患者,可能会出现高血压眼底改变、心功能不全、慢性肾功能不全、肾衰竭、脑梗死或脑出血等并发症,严重影响患者的预后及生活质量。
(一)糖皮质激素
糖皮质激素是17-OHD最主要的治疗药物,且为终身治疗。成人多使用地塞米松,初始剂量为0.75~2.0mg/d,维持剂量为0.1~0.375mg/d,具体剂量可根据患者体重、血压、血钾等变化进行调整。一般血钾在服药1周内恢复正常,血压在服药1~4周降至正常或达到正常的高限。少部分患者单用糖皮质激素血压控制不佳,需要加用盐皮质激素受体拮抗剂如螺内酯等药物辅助治疗。
(二)盐皮质激素受体拮抗剂和降压药物
最常用的药物是螺内酯,剂量为50~200mg/d,分1~2次服用。根据血压、DOC和电解质的监测结果,逐渐调整药物剂量。若存在明显不良反应,可选择依普利酮治疗。保钾利尿剂如阿米洛利对低钾血症非常有效,但在控制血压方面不如螺内酯。降压药物可选用钙离子通道阻滞剂来辅助控制血压。
(三)性激素替代治疗
可采用小剂量雌激素诱导青春期的出现。选择女性表型的患者,予以雌激素替代治疗以促进女性第二性征发育。一般从青春期开始使用,成人确诊后即可使用。子宫发育完整的46,XX女性患者可择期予以孕激素治疗,进行孕酮撤退诱导月经来潮。极少数核型为46,XY的男性患者选择以男性性别抚养和生活,因睾酮分泌不足,需要雄激素替代治疗。
(四)手术治疗
对于阴道发育较差的女性患者,根据性心理需求,择期行阴道成形术。少数女性患者伴有卵巢囊肿、囊肿破裂或伴明显疼痛症状时需手术治疗。核型为46,XY的男性患者因发育不良的异位睾丸组织恶变率高,如选择以女性性别生活,需手术切除异位睾丸。核型为46,XY的部分型17-OHD男性患者需外科手术治疗尿道下裂;或者对于选择按照女性生活的患者,需考虑“阴蒂”缩小手术。









