


 收藏
收藏 已收藏
已收藏1967年Refetoff首先介绍了甲状腺激素不敏感综合征,它的基本特征是高甲状腺激素血症,TSH水平不适当地升高或保持正常。甲状腺激素(TH)的生物学作用至少包括了6个单独的步骤,其中有三个步骤与甲状腺激素不敏感有关。首先被临床认识的是T3Rβ突变,目前已经有1000例以上的报道,来源于300多个家系,占甲状腺激素不敏感综合征的85%左右;少数甲状腺激素不敏感综合征是甲状腺激素的细胞膜转运蛋白MCT8突变或甲状腺激素的细胞内代谢因子SECISBP2(含硒)突变所致[1]。
由于不敏感的组织细胞不同、缺陷严重程度和代偿的程度不同,甲状腺激素不敏感综合的临床表现极不均一,可从无任何症状到症状极为严重。本综合征有家族发病倾向,少数为散发性[1-4]。发病年龄多从婴儿期开始,但症状轻者也有到老年始获诊断者。有些病例表现为甲亢或甲减,故常被误诊,甚至采取不适当的治疗措施。目前,已报道的甲状腺激素不敏感综合征病例有600余例,鉴定出的甲状腺激素受体(T3R)基因突变点有100多个。
(一)T3受体基因突变导致的甲状腺激素抵抗
T3R有T3Rα和T3Rβ两种异构体,T3Rα有 T3Rα1和 T3Rα2两种亚型,T3Rβ有T3Rβ1和T3Rβ2两种亚型,四种亚型在不同组织的分布、性质、作用均有一定差异。T3Rα1、T3Rα2和T3Rβ1mRNA在所有的组织都能表达,而高浓度的T3Rβ2mRNA只在腺垂体TSH分泌细胞中表达。T3Rα2不能与T3高亲和力结合。患者的T3R基因的两个等位基因中有一个异常,突变可为家族性或散发性,散发性病例的病因可能还有其他因素的参与[5,6]。
1.突变型T 3受体
THRA与THRB分别编码甲状腺激素受体TRα1和TRβ1/2,THRB基因突变引起血清甲状腺激素和TSH均升高,THRA基因突变者的血清T4相对较低而T3升高,伴有生长发育障碍和骨龄延迟。
一般为常染色体显性或隐性遗传;偶尔,胚胎早期的嵌合性突变仅发生于某些组织的某些细胞系[2]。T3R基因突变可导致T3R结构和功能异常,引起甲状腺激素的作用障碍。目前报道的T3R基因突变以点突变最多,集中在T3Rβ亚基的激素结合区,如 A229T、M305T、A312T、R315C、R315H、D317H、G327R、L330S、R333W、Q335H、R338L、R338W、G340N、G340R、G342E、G342Q、G345S、T426I、R429Q、R433H、S437G、M437V、K438E、L445H、P446I、F448T、F448S、P453S、F454C、V458A和M928L等[7]。1例患儿及家系成员进行TRβ基因突变位点分析发现患儿及其母亲TRβ基因第9外显子突变,第1235位碱基由胞嘧啶变成腺嘌呤,导致该位点编码的氨基酸由丙氨酸变为天冬氨酸,发现新的中国人家系TRβ基因突变,但患儿母亲没有相应的临床表现[8]。
T3Rβ亚基基因突变后,耳蜗组织表达的T3Rα1不能代偿T3Rβ的生理作用,从而导致耳聋。DFNB4基因突变引起常染色体隐性遗传性非Pendred综合征性耳聋(感觉神经性失聪),与pendred基因都定位于7q,说明内耳的发育与T3Rβ功能有密切联系。
突变型T3R基因所表达的T3R功能异常,对T3的亲和力降低。突变型T3Rβ不能或减少与T3的结合或不能形成二聚体,因此与DNA结合减少。另外,突变型T3Rβ还可与野生型T3R竞争T3结合位点而抑制野生型T3Rβ的功能,或与野生型或突变型T3Rβ分别形成异二聚体和同二聚体,从而减少了与DNA的结合。根据对患者T3Rβ基因的分析,患者两个T3Rβ等位基因中的一个有突变,另一个正常。
2.突变型T3受体作用机制
当两个等位基因都存在突变(或丢失)时,突变的T3R不能正常地执行其功能,通常以优势负性作用(dominant negative effect)方式起作用。在这种方式下,即使存在正常的受体,甲状腺激素的生物作用也是下降的。但突变的T3R仍能与一个正常的T3R以同二聚体结合到T3R反应元件(TRE)上,这一复合物还能吸引辅抑制子,但因T3不能与其结合,故在TRE区域上仍存在一个稳定的非活化复合物。当残余的T3被结合后,同二聚体解离,突变型T3R与视黄酸样X受体或其他辅助因子结合。在这种情况下,突变受体发生异二聚化,然后与TRE结合,但是阻断了含有辅活化子的异二聚体的正常T3R进入其内,从而导致基因的失活。因此,最常发生的变化是激素结合反应丧失,T3不能与T3R结合,T3R不能释放辅抑制子,基因无法被激活。虽然有些突变型T3R还能结合T3,但T3R释放辅抑制子的速度缓慢。此外,还有人提出一种机制是由于突变型T3R分子形成异二聚体并返回到TRE上,但不能吸引辅活化子,因而靶基因处于不表达状态;或者,因T3R突变,选择性地抑制了β2二聚体的功能。
3.临床表现
症状不一致与不同组织的T3受体亚型及辅抑制子/辅激活子的活性差异有关。有人认为,全身性和部分性T3R缺陷可能是同一基因病谱中的两种不同表现,症状多样化的原因是由于各组织中的T3R表达水平不同;当然,症状的不一致也与不同组织的T3R亚型分布不同有关。例如,心脏富含T3Rα,由于T3R不敏感综合征患者的T3Rα正常,但血清FT3升高,加上特定组织中的辅抑制子/辅激活子的活性存在差异,所以同样的突变在同一个家庭中被诊断为部分性甲状腺激素不敏感,而在另一个家庭中却表现为全身性甲状腺激素不敏感,甚至两种类型可同时出现在同一家族中。
全身性甲状腺激素抵抗(generalized resistance of thyroid hormone,GRTH)多为常染色体显性遗传,其病因与人 C-erbAβ基因(编码T3Rβ亚基)有关(位于第3号染色体上)。
(二)其他基因突变导致的甲状腺激素抵抗
1.T 3R辅激活与突变
有些甲状腺激素不敏感综合征并无T3Rα或T3Rβ基因序列突变。在65个甲状腺激素不敏感综合征家族中,有6个家族的T3Rβ1和T3Rβ2均无异常,但这些患者的临床表现与一般T3R突变者相似。进一步研究发现,这些患者是由于辅激活子(coactivator,如 NcoA-1或NcoA-3),或辅抑制子(如 NcoR、SMRT等),或辅调节子(如RXRr等)的突变或其他因子的突变所致。
2.MCT8突变
少数甲状腺激素不敏感综合征是甲状腺激素的细胞膜转运蛋白MCT8突变所致,患者伴有严重的精神运动障碍,目前的病例大约来源于20多个家系的100多个男性患儿。此外,目前有4例患者是由于甲状腺激素的细胞内代谢因子SECISBP2(含硒)突变所致,来源于2个家系;SECISBP2是合成含硒蛋白(selenoprotein,如脱碘酶)的必需因子。此外,有的患者T3R结合T3最大容量可减少到只有正常人的10%~65%。胱氨酸血症可引起继发性垂体型甲状腺激素不敏感综合征,而个别病例的病因仍未明了。
MCT8是脑组织的一种T3转运体。MCT8基因突变导致X-性连锁性 MCT8缺陷症(Allan-Herndon-Dudley综合征)。临床上以男性发育障碍、精神运动型发育延迟和肌张力低下为特征,患儿肌肉关节松软,颈部松软,不能竖头或站立,深肌腱反射缺乏。MRI显示脑白质髓鞘形成缺陷或髓鞘化不足[9-16],与Pelizaeus-Merzbacher病(PLP1突变)的表现相似。但Allan-Herndon-Dudley综合征的轴向肌张力减退持续存在,四肢由肌张力减退可进展为肌痉挛和椎体外症状和智力障碍[17-23]。
3.特殊表现
血清T3升高,脑MRI可发现髓鞘形成延迟(delayed myelination)。新生儿期血清甲状腺激素的变化与T3受体α1(T3Rα1)突变相似。血清T4正常或轻度降低,TSH正常或轻度升高,T3显著升高[24-28]。但是新生儿期过后,AHDS患儿发生典型甲减表现,如生长发育延迟、骨骼发育不良、肌肉张力减退、便秘等,而认知功能相对完好[29,30]。
甲状腺激素替代治疗对症状无效,显著升高的血清T3可被硫脲类药物或二碘甲腺丙酸(diiodothyropropionic acid)抑制,但对运动和认知功能无帮助。着力强调运动康复和对症处理。如果母亲携带突变型SLC16A2基因,男性子代的患病和女性子代携带致病基因的风险为50%。
根据天然与人工合成的甲状腺激素制剂的代谢特点(表6)、疾病的严重程度和不同类型做出治疗决策,且应维持终生。轻型无症状者可不予治疗。
表6 天然与人工合成的甲状腺激素制剂的代谢特点
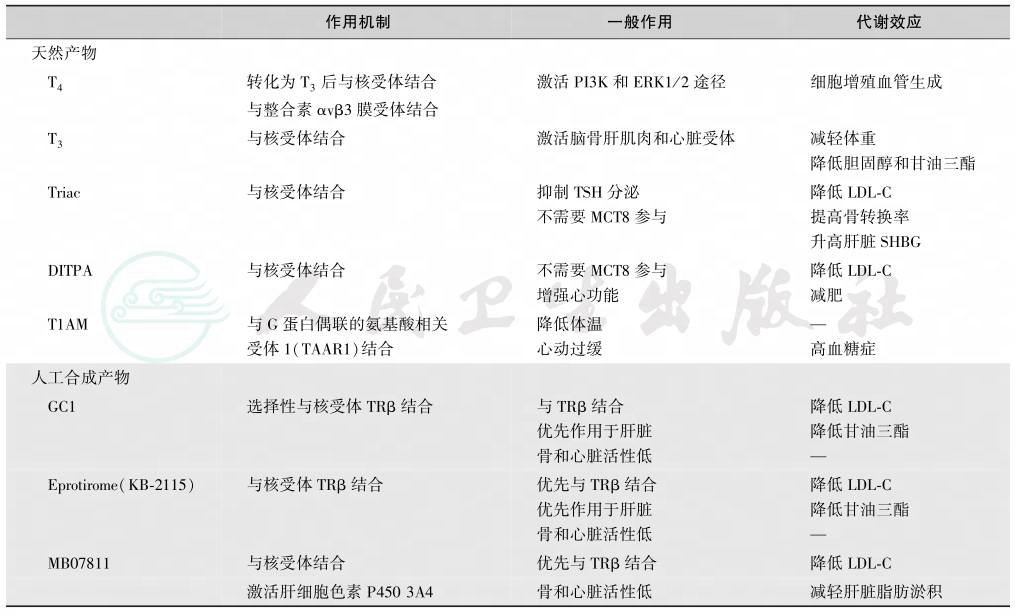
(一)甲状腺激素不敏感综合征治疗
不论何种类型的甲状腺激素不敏感综合征均可采用L-T3治疗(25μg/d)。选择性垂体不敏感型尽管血TT3和TT4升高,但用T3治疗不仅不使患者的甲亢加重,相反由于血T3更加升高,反馈抑制了垂体TSH分泌,可使血清TSH逐渐降低到正常,血清甲状腺激素也随之降低,甲状腺缩小,甲亢症状得到改善或消失。但L-T4治疗无效,因此有人提出此型患者可能T4转变为T3有缺陷,但未得到证实。最近报道,D-T3可收到类似治疗效果,但机制未明。全身型和周围型甲状腺激素不敏感综合征的剂量大小应个体化。
Redetti等报道,用三碘甲腺乙酸(triiodothyroacetic acid,TRIAC)治疗垂体型甲状腺激素不敏感综合征患者2年,未发现明显不良反应,甲亢症状控制良好。有的周围型甲状腺激素不敏感患者,在每天给予250~500μg的L-T4才使部分指标恢复正常。全身型者用L-T4治疗后,血清TSH可降低,甲减症状得到改善。对婴幼儿发病者治疗应尽早实施,否则严重病例可有生长发育延迟和脑发育障碍。
(二)选择性垂体甲状腺激素不敏感综合征的治疗
溴隐亭用于治疗选择性垂体不敏感型患者,可使血TSH降低。从小剂量开始,逐渐加量,使血清TSH和甲状腺激素恢复正常,甲亢症状随之消失。长期疗效如何,尚待进一步观察。其他可抑制TSH分泌的药物有地塞米松和生长抑素及其类似物。但此两药长期应用不良反应大,因此难以用作长期治疗本型患者的药物。
(三)伴甲亢的甲状腺激素不敏感综合征治疗
对所有患者禁用治疗甲亢的方法处理,如抗甲状腺药、核素碘(131I)和手术切除甲状腺等,因为这些治疗方法不仅无效,而且对婴幼儿患者可造成不可逆性脑与骨的损害,且使临床症状加重。垂体不敏感型者TSH分泌已经增多,上述治疗使血清甲状腺激素降低,对垂体负反馈作用进一步减弱,因此可导致垂体TSH细胞增生或TSH瘤。如果由于误诊而行手术或放射性碘治疗,导致甲减,则应给予甲状腺激素替代治疗,使血清TSH降至正常范围内。
1.Refetoff S,Dumitrescu AM.Syndromes of reduced sensitivity to thyroid hormone:genetic defects in hormone receptors,cell transporters and deiodination.Best Pract Res Clin Endocrinol Metab,2007,21(2):277-305.
2.Mamanasiri S,Yesil S,Dumitrescu AM,et al.Mosaicism of a Thyroid Hormone Receptor(TR)Beta Gene Mutation in Resistance to Thyroid Hormone(RTH).J Clin Endocrinol Metab,2006,91:3471-3477.
3.Brent GA.Mechanisms of thyroid hormone action.J Clin Invest,2012,122(9):3035-3043.
4.Onigata K,Szinnai G.Resistance to thyroid hormone.Endocr Dev,2014,26:118-129.
5.van Mullem AA,Visser TJ,Peeters RP.Clinical Consequences of Mutations in Thyroid Hormone Receptor-α1.Eur Thyroid J,2014,3(1):17-24.
6.Schoenmakers N,Moran C,Peeters RP,et al.Chatterjee K.Resistance to thyroid hormone mediated by defective thyroid hormone 7.Visser TJ.Thyroid hormone transporters and resistance.Endocr Dev,2013,24:1-10.
7.俞放,赵咏桔,陈瑛,等.甲状腺激素受体β基因V458A点突变所致甲状腺激素抵抗综合征.中华内分泌代谢杂志,2004,20(4):311-313.
8.Dumitrescu AM,Refetoff S.The syndromes of reduced sensitivity to thyroid hormone.Biochim Biophys Acta,2013,1830(7):3987-4003.
9.Bernal J,Guadaño-Ferraz A,Morte B.Perspectives in the study of thyroid hormone action on brain development and function.Thyroid,2003,13:1005-1012.
10.Morreale de Escobar G,Obregon MJ.Escobar del Rey F.Role of thyroid hormone during early brain development.Eur J Endocrinol,2004,151:U25-U37.
11.Heuer H,Visser TJ.Minireview:pathophysiological importance of thyroid hormone transporters.Endocrinology,2009,150:1078-1083.
12.Visser WE,Friesema EC,Visser TJ.Minireview:thyroid hormone transporters:the knowns and the unknowns.Mol Endocrinol,2011,25:1-14.
13.Kersseboom S,Visser TJ.Tissue-specific effects of mutations in the thyroid hormone transporter MCT8.Arq Bras Endocrinol Metabol,2011,55:1-5.
14.Ceballos A,Belinchon MM,Sanchez-Mendoza E,et al.Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-L-thyronine.Endocrinology,2009,150:2491-2496.
15.Wirth EK,Roth S,Blechschmidt C,et al.Neuronal 3’,3,5-tri-iodothyronine(T3)uptake and behavioral phenotype of mice deficient in Mct8,the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome.J Neurosci,2009,29:9439-9449.
16.Dumitrescu AM,Liao XH,Best TB,et al.A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene.Am J Hum Genet,2004,74:168-175.
17.Friesema ECH,Grueters A,Biebermann H,et al.Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation.Lancet,2004,364:1435-1437.
18.Allan W,Herndon CN,Dudley FC.Some examples of the inheritance of mental deficiency:apparently sex-linkediodicy and microcephaly.Am J Ment Defic,1944,48:325-334.
19.Lóez-Marín L,Martín-Belinchón M,et al.MCT8-specific thyroid hormone cell transporter deficiency:a case report and review of the literature.Rev Neurol,2013,56:615-622.
20.Jansen J,Friesema EC,Kester MH,et al.Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8.Endocrinology,2008,149:2184-2190.
21.Schwartz CE,Stevenson RE.The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome.Best Pract Res Clin Endocrinol Metab,2007,21:307-321.
22.Tonduti D,Vanderver A,Berardinelli A,et al.MCT8 deficiency:extrapyramidal symptoms and delayed myelination as prominent features.J Child Neurol,2013,28:795-800.
23.van der Knaap MS,Wolf NI.Hypomyelination versus delayed myelination.Ann Neurol,2010,68:115.
24.Gika AD,Siddiqui A,Hulse AJ,Edward S,et al.White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene.Dev Med Child Neurol,2010,52:475-482.
25.Filho HC,Marui S,Manna TD,et al.Novel mutation in MCT8 gene in a Brazilian boy with thyroid hormone resistance and severe neurologic abnormalities.Arq Bras Endocrinol Metabol,2011,55:60-66.
26.Bochukova E,Schoenmakers N,Agostini M,et al.A Mutation in the thyroid hormone receptor alpha gene.N Engl J Med,2012,366:243-249.
27.Zug A,Visser TJ,Uitterlinden AG,et al.A child with a deletion in the monocarboxylate transporter 8 gene:7-year follow-up and effects of thyroid hormone treatment.Eur J Endocrinol,2011,165:823-830.
28.Wémeau JL,Pigeyre M,Proust-Lemoine E,et al.Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8.J Clin Endocrinol Metab,2008,93:2084-2088.
29.Verge CF,Konrad D,Cohen M,et al.Diiodothyropropionic Acid(DITPA)in the treatment of MCT8 deficiency.J Clin Endocrinol Metab,2012,97:4515-4523.
30.Dumitrescu AM,Fu J,Dempsey MA,et al.MCT8-Specific Thyroid Hormone Cell-Membrane Transporter Deficiency.In GeneReviewsTM Edited by Pagon RA,Adam MP,Bird TD.Seattle(WA):University of Washington,Seattle,2010,1993-2013.http://www.webcitation.org/query.php?url=http://www.ncbi.nlm.nih.gov/books/NBK26373/&refdoi=10.1186/1471-2431-14-252.
31.Refetoff S.Syndrome of thyroid hormone resistance.Am J physiol,1982,243:E88-E98.
32.胡志兵,胡毅.甲状腺激素不敏感综合症一例.中华内分泌代谢杂志,1995,11(1):60.
33.吴研,张爱珍.甲状腺激素不敏感综合征三例报道及文献复习.中华内分泌代谢杂志,2011,27(6):523-524.
34.Refetoff S.Resistance to thyrotropin.J Endocrinol Invest, 2003, 26(8):770-779.









