


 收藏
收藏 已收藏
已收藏英文名称 :Thyrotoxicosis and Graves disease
甲状腺毒症(thyrotoxicosis)是任何原因引起血液循环中甲状腺激素(thyroid hormones,TH)过多,引起以神经、循环、消化等系统兴奋性增高和代谢亢进为主要表现的一组临床情况的总称;而甲状腺功能亢进症(hyperthyroidism,简称甲亢)是指产生和分泌TH过多和甲状腺功能过高引起的一组临床综合征。甲状腺毒症包括了甲亢,而甲亢只是甲状腺毒症中的一种类型。TH过量所致的临床状态称为甲状腺毒症较甲状腺功能亢进症确切,因为TH过量既可来源于甲状腺病变,又可由非甲状腺疾病甚至含TH的药物引起。由于临床习惯的关系,本书仍应用“甲亢”一词;但严格地说,不能将甲状腺毒症和甲亢等同称呼。
甲亢的病因复杂(表1),以Graves病(Graves disease,GD)最常见,约占所有甲亢患者的85%。多见于成年女性,男性与女性比为1∶4~1∶6。GD亦称弥漫性毒性甲状腺肿(diffuse toxic goiter),在欧洲多称为Basedow病或Parry病;在我国、美洲和其他地区常称为GD或弥漫性毒性甲状腺肿。典型病例除有甲状腺肿大和高代谢症群外,尚伴有不同程度的眼病,少数有眼病而不伴甲亢(5%),但存在甲状腺免疫功能紊乱或其他实验室检查异常,称为甲状腺功能“正常”的眼病(euthyroid Graves ophthalmopathy,EGO)。单眼受累占10%~20%,少数(5%)患者可有皮肤病变(胫前黏液性水肿及指端粗厚等)或肌病。从病理上看,GD具有自身免疫性甲状腺炎的某些特征,个别慢性淋巴细胞性甲状腺炎可演变为GD,反之亦然;因而亦有人将GD归为自身免疫性甲状腺炎(autoimmune thyroiditis,即3型自身免疫性甲状腺炎。但是,更普遍的观点认为,GD是一种伴TH分泌增多的特殊自身免疫性甲状腺病(autoimmune thyroid disease,AITD)。本文主要介绍GD。
表1 甲亢的病因分类

GD的发病机制和病因未明。GD是 AITD中的一种;一般认为,GD以遗传易患性为背景,在感染、精神创伤等因素作用下,诱发体内的免疫系统功能紊乱,免疫耐受、识别和调节功能减退和抗原特异或非特异性抑制性T细胞(Ts)功能缺陷,机体不能控制针对自身组织的免疫反应,Ts细胞减弱了对辅助T细胞(Th)的抑制,特异B淋巴细胞在特异Th细胞辅助下产生异质性免疫球蛋白(自身抗体)。同时,存在缺陷的Ts功能降低、Th不适当致敏和IL-1和IL-2等的参与是B淋巴细胞产生大量自身抗体的重要原因。甲状腺自身组织抗原主要有TSH、TSH受体、甲状腺球蛋白(Tg)、甲状腺过氧化物酶(TPO)及Na+/I-同转运蛋白(NIS)等。
(一)GD是特殊的自身免疫性甲状腺病
1.甲状腺自身抗体
最初,这类自身免疫性物质被称为长效甲状腺刺激物(long-acting thyroid stimulator,LATS),以后由于测定方法不同,分别称为人甲状腺刺激物(human thyroid stimulator,HTS)、LATS保护物(LATS protector,LATSP)、TSH置换活性(thyrotropin displacement activity,TDA)、甲状腺刺激免疫球蛋白(thyroid stimulating immunoglobulin,TSI)、甲状腺刺激性抗体(thyroid-stimulating antibody,TSAb)或TSH受体抗体(thyrotropin receptor antibodies,TRAb)等。现已明确,TRAb是淋巴细胞分泌的一组多克隆抗体,可与TSH受体的不同位点结合。
TRAb至少分为3类。在兴奋型抗体中,有一类与TSH受体结合后,促进TH合成和释放,甲状腺细胞受刺激而增生,称为甲状腺刺激性(兴奋性)抗体(thyroid stimulating antibody,TSAb),TSAb为自身抗体的主要成分;另一类与TSH受体结合后,仅促进甲状腺细胞肿大,不促进TH的合成和释放,称为甲状腺生长刺激免疫球蛋白(thyroid growth immunoglobulin,TGI);封闭型自身抗体与TSH受体结合后,阻断及抑制甲状腺功能,称为甲状腺功能抑制抗体(thyroid function inhibitory antibodies,TFIA)或甲状腺生长封闭性抗体(thyroid growth blocking antibodies,TGBAb)。少数GD患者虽有明显的高代谢症群,但甲状腺肿大甚轻微,可能是由于体内的兴奋性抗体(TSAb)占优势所致。在正常情况下,TSH与TSHR的细胞外区域相结合,激活刺激性G蛋白α亚单位(GSα),使腺苷酸环化酶活化,发挥与TSH相同的生物效应;TSHR激活G蛋白q亚单位,导致磷脂酶C活化,使甘油二酯(diglyceride,DG)及三磷酸肌醇(IP3)增多。TSHR还通过结合到其他G蛋白受体家族的成员发挥作用。
2.细胞免疫功能异常
用单独的免疫监视缺陷尚不能解释某些特异的免疫反应,这些特异性免疫反应与免疫信号的独特型级联反应(idiotype cascade)有关。在免疫球蛋白分子中,重链和轻链的可变区具有抗原决定簇,根据该区氨基酸序列决定抗体的特异性,不同特异性的可变区具有不同的抗原决定簇或基因型决定簇。例如,家兔以单克隆人骨髓瘤蛋白免疫所得的抗血清可与骨髓瘤免疫球蛋白的可变区特异性结合,这种结构称为基因型。将独特型(基因型)/抗独特型(idiotye/antiidiotype)原理扩大应用,可解释GD和重症肌无力等疾病的受体抗体的形成。如在重症肌无力,配体(乙酰胆碱)能和细胞表面受体及其相应抗体(抗-乙酰胆碱抗体,即基因型)相结合,因而受体和抗体具有相同的可与配体结合的分子结构。同理,Farid等以抗人TSH抗体免疫家兔可获得抗基因型抗体,后者在甲状腺培养细胞中既能和TSH受体结合,也能刺激cAMP合成,故其生物学行为与GD中的LATS或TSAb并无不同。
此外,本病中针对甲状腺组织的白细胞移动抑制试验呈阳性反应,甲状腺和眼球后组织均有明显的淋巴细胞浸润,说明还有细胞介导的免疫反应参与。细胞间黏附分子(intercellular adhesion molecule-1,ICAM-1)、白细胞功能相关抗原(leukocyte functional associated antigen-1,LFAA)、与内皮细胞有关的黏附分子也与GD发病有关。
(二)遗传素质/感染/精神因素参与GD发病
1.遗传因素
(1)家族因素
部分GD有家族史,同卵双生相继发生GD者达30%~60%,异卵双生仅为3%~9%。流行病学调查发现,GD亲属中患另一种AITD(如慢性淋巴细胞性甲状腺炎)的比例和TSAb的检出率均高于一般人群,但GD究竟以何种方式遗传仍不清楚。
(2)HLA抗原
与GD遗传易患性有关的基因分为人类白细胞膜组织相容性抗原(HLA)和非HLA候选基因两大类。HLADR3与GD的相关性已在白种人中得到证实,HLA A2和DBP1*0501在促甲状腺素结合抑制免疫球蛋白(TBⅡ)阳性的GD中出现频率增加。在TBⅡ阴性的GD中,HLA B46和DBP1*0202共显率较高。高加索人中的HLA-B8、日本人中的HLA-B35和中国人中的HLA-BW46都是GD的相对危险因子。
(3)非HLA抗原
TSHR基因、干扰素-γ基因、肿瘤坏死因子-β基因、白细胞介素-1受体拮抗剂基因、IL-4基因、T3R-β基因、T细胞抗原受体基因、热休克蛋白70基因、补体C4基因和维生素D受体基因都与GD遗传易患性有关的非HLA候选基因,但尚无一种遗传学指标能够准确预测GD的发生。
(4)T淋巴细胞抗原
细胞毒性T淋巴细胞抗原4(CTLA4)基因可能是影响GD遗传易患性的主要非HLA候选基因之一。CTLA 4基因中Thr17A1a多态性中的G等位基因(AG)与GD相关,CTLA4和HLA基因位点的共同作用可能占GD遗传易患性的50%。另外,CTLA4基因17密码子双等位基因多态性(AG)与甲状腺相关性眼病(thyroidassociated ophthalmopathy,TAO)的易患性也有关。CTLA4基因启动子的变异体降钙素(calcitonin,CT)是GD的另一个危险因素。
2.感染因素
细菌或病毒可通过3种可能途径启动AITD发病:①分子模拟(molecular mimicry),感染因子和TSH受体间在抗原决定簇方面的相似分子结构,引起抗体对自身TSH受体的交叉反应,例如在耶尔森肠杆菌(Yersinia enterocolitica)中,具有TSH受体样物质能增加甲亢发病的危险性。②感染因子直接作用于甲状腺和T淋巴细胞,通过细胞因子诱导Ⅱ类MHC,HLA-DR在甲状腺细胞表达,向T淋巴细胞提供自身抗原作为免疫反应对象。③感染因子产生超抗原(superantigen)分子,诱导T淋巴细胞对自身组织起反应。
由环境因素引起AITD的典型例子是病毒和细菌感染,如耶尔森菌性肠炎、柯萨奇B病毒感染、反转录病毒感染或幽门螺杆菌感染等,而感染与AITD关系最密切者是丙型肝炎。丙型肝炎病毒可进入甲状腺滤泡细胞,即使不直接损伤甲状腺,病毒蛋白也可以产生深远的免疫反应。例如,丙型肝炎E2蛋白可诱导甲状腺细胞凋亡,上调前炎症因子表达并激发甲状腺自身免疫反应。干扰素激活JAK-STAT通路和干扰素刺激基因(interferon-stimulated genes),增加MHC I类抗原表达,使免疫反应转向Th1模式,并进一步引起干扰素γ与IL-2分泌,增强免疫淋巴细胞、巨噬细胞和NK细胞的活性、活化中性粒细胞与单核细胞。此外,IFN还对甲状腺有毒性作用,引起细胞凋亡和免疫反应。
3.精神因素
部分GD患者在临床症状出现前有明显的精神刺激或创伤史,精神因素引起GD很可能是通过免疫系统发生的。有人认为,精神创伤后出现的GD是通过中枢神经系统作用于免疫系统,精神因素使中枢神经系统去甲肾上腺素降低,CRH和ACTH及皮质醇分泌增多,免疫监视功能降低,B淋巴细胞增生,分泌的TSAb增多,进而引起GD。也有人认为,因GD起病缓慢,精神创伤后突然发病可能是原有疾病突然加重才开始引起注意,因而认为精神因素并非直接的致病原因。
(三)GD眼病与遗传/环境/免疫因素有关
甲状腺相关性眼病(TAO)主要包括非浸润性突眼和浸润性突眼。一般认为,非浸润性突眼与TH增多所致的交感神经兴奋性和眼肌紧张性增高有关,而浸润性突眼是眶后组织自身免疫炎症的一种表现。TAO的发病有多种因素参与,可能与遗传因素、环境因素(如吸烟)和免疫等因素均有关。
1.遗传因素
GD眼病的遗传因素可能涉及多个基因,目前提出了50多个相关基因,其中可能以HLAⅡ型、CTLA-4、PTPN22、CD40、Tg和TSHR最为重要。虽然HLA B8、DR3与TAO相关,但目前并没有发现某个特定的基因能引起TAO遗传易患性的改变。有人认为,TNFα基因5’端的多态性与GD眼病的发病有关。另有人发现,TAO与P1血型有关,TAO患者P1阳性较GD不伴眼病者多见。
2.环境因素
吸烟是一个重要的危险因素。吸烟降低硫脲类药物控制甲亢的缓解率,改变TSH受体的免疫原性,诱发TSH受体刺激性抗体的生成,后者与眼球后组织的TSH受体作用,加重免疫反应。此外,吸烟还与T/B淋巴细胞的免疫功能有关,吸烟伴随的缺氧也是Graves 病和突眼的重要原因。有人观察到,83%的TAO是吸烟者,吸烟者的TAO较非吸烟者严重。而吸烟者的TAO行糖皮质激素和眶部放疗后,改善程度较非吸烟者小。其可能原因为:①尼古丁或焦油刺激成纤维细胞中的HLA-DR抗原表达,粘多糖产生增加;②成纤维细胞在低氧的情况下合成更多的粘多糖;③吸烟者体内IL-1受体拮抗物的浓度较非吸烟者低,这种改变引起TAO对糖皮质激素和放疗反应不敏感;④吸烟者中,热休克蛋白-72(heat shock protein,HSP-72)也在眼眶成纤维细胞上表达。
许多药物(如GH、胰岛素或促进眼压升高的药物等)对TAO有明显影响。一些GD患者在接受131I治疗后,TAO恶化,其原因未明。另据报道,毒性甲状腺结节在使用131I治疗后亦可发生TAO。
一般来说,AITD在妊娠期有不同程度改善,而在分娩后病情加重,或发生新的AITD;这种现象与妊娠期的免疫功能变化有关。因此,妊娠后数年内妇女发生GD的风险明显增加。此外,眼部手术,尤其是白内障手术可能诱发或恶化TAO,因此眼部手术前需认真评价手术风险,术后应严密观察TAO的变化。其他提示复发的因素包括:①年轻男性;②具有AITD家族史;③吸烟;④突眼;⑤T3/T4比值升高;⑥治疗后形成甲状腺结节或甲状腺超声显示低回声。
3.体液免疫因素
20世纪80年代,有人在TAO患者血清中查到抗可溶性眼外肌抗原的循环自身抗体及抗球后结缔组织抗体。随后发现抗眼外肌膜的64kD蛋白及抗成纤维细胞的23kD蛋白,33%的TAO患者64kD抗体阳性,病程短于12个月者阳性率达75%,而正常人均为阴性。甲状腺和眼球后组织存在共同抗原,产生的交叉免疫反应可导致GD的甲状腺外组织病变。用PCR方法扩增特异的cDNA,发现成熟TSH-RmRNA在眼球后组织表达;GD患者眼球后成纤维细胞上存在有与该抗血清反应的蛋白质,分子量分别为95kD、71kD和18kD。最近发现,突眼与TSAb升高有关,而与眼外肌受累无关,TAO与抗眼肌抗体有一定关系。
4.细胞免疫因素
Gunji等报道,编码Gαs蛋白的cDNA(411bp)开放阅读框架相当于含121个氨基酸残基的蛋白质(1.4kb)及另一种与Gαs无同源序列的蛋白质(基因0.3kb)。Gαs在甲状腺和眼肌中表达,这两种组织中存在的抗体结合的Gαs免疫反应性(抗原决定簇)可能与TAO的发生有关。研究发现,眼外肌的肌内膜成纤维细胞及球后结缔组织中的成纤维细胞有HLA-DR及ICAM-1表达。TAO患者的眶部及眶前成纤维细胞表达HSP-72。在GD及慢性淋巴细胞性甲状腺炎患者中,甲状腺滤泡细胞表面也可检出HSP-72的表达。现认为,HLA-DR涉及T淋巴细胞抗原识别,ICAM在结缔组织细胞与免疫活性细胞相互作用中起作用,HSP-72可增加细胞间的相互作用,与HLA-DR分子结合,递呈抗原给T细胞,因此推测眶部浸润的单核细胞攻击组织可能依赖于这些免疫调节蛋白的相互作用及协调表达。眼外肌、脂肪细胞、炎症浸润细胞中的IGF-1也可能与TAO的发病有关。成纤维细胞活性增强与突眼有密切关系,使粘多糖、胶原、糖蛋白分泌增多,特别是粘多糖有较强的吸水性,进而使脂肪组织、眼外肌间质水肿。
一些学者发现GD患者的淋巴细胞对眶部提取物可产生移动抑制因子(migration inhibitory factor,MIF)。Wang等用51Cr释放法检测发现,TAO患者存在对眼肌细胞的抗体依赖性细胞介导的细胞毒(antibody-dependent cell-mediated cytotoxicity,ADCC)作用,但对成纤维细胞无ADCC作用,且眼肌细胞溶解的百分率与眼肌受累和眶部炎症的程度相关,但未观察到抗眼肌细胞的抗体依赖性补体介导的细胞毒(antibody-dependent complement-mediated cytotoxicity,AMC)作用。
早期眼球后组织以细胞免疫反应为主,随着病情的发展,转为体液免疫起主导作用。T淋巴细胞仅在活动性TAO早期明显,但成纤维细胞表达的HLA-DR抗原增多在所有阶段都可观察到,也表明T淋巴细胞更多地参与疾病的早期过程。浸润TAO患者球后的淋巴细胞大多是近期激活的T淋巴细胞以及一些记忆T淋巴细胞。自身免疫反应性T淋巴细胞(CD4+)识别甲状腺、眶内组织及眼球外自身抗原并与其受体结合而被激活,产生各种黏附分子和细胞因子,并激活CD8+T淋巴细胞或B细胞,产生各种自身抗体。眶周浸润的T淋巴细胞按其基因型可主要分为分泌IL-1、IL-6、IFN-γ、TNF-α的Th1型及表达分泌IL-4、5、10的Th2型。IFN-γ、IL-1α是球后成纤维细胞产生葡糖胺聚糖(glycosaminoglycan,GAG)的强烈刺激因子,这些细胞因子还刺激成纤维细胞增殖,分化为成熟脂肪细胞,使眶后脂肪组织容量增加,最终导致突眼,但可被糖皮质激素阻断。有学者发现,眼肌肥厚与眼肌内TNF-αmRNA及IL-10mRNA表达有关,眼眶体积与IL-6mRNA表达呈正相关,与IL-4mRNA表达呈负相关。
(一)滤泡增生肥大伴淋巴细胞浸润是GD的病理特征
GD的甲状腺呈对称性、弥漫性增大,甲状腺内血管增生,血运丰富,使甲状腺外观为红色。滤泡细胞增生肥大,细胞呈立方或柱状,滤泡细胞由于过度增生而形成乳头状折叠凸入滤泡腔内,高尔基复合体肥大,附近有许多囊泡,内质网发育良好,核糖体和线粒体增多。滤泡腔内胶质减少甚或消失。甲状腺内可有淋巴细胞浸润,或形成淋巴滤泡,或出现淋巴组织生发中心。
经治疗后甲状腺的形态结构可发生相应的变化。短期使用大剂量碘剂后,甲状腺可迅速缩小,腺泡中胶质含量增多,滤泡细胞变为立方状或扁平状,乳头状结构消失,血管减少。长时间使用硫脲类抗甲状腺药物后,可使甲状腺组织呈退行性改变,滤泡增大富含胶质,大部分滤泡细胞呈扁平或矮立方形,少部分滤泡细胞仍肥大,或可见到上皮嵴及短小乳头状结构。此时活检标本不易与甲状腺肿鉴别。并发甲状腺内出血罕见。
(二)球后脂肪细胞和免疫细胞浸润-水肿是GD眼病的突出特点
GD仅有良性眼病时常无异常病理改变。在浸润性突眼患者中,球后组织中常有脂肪浸润,脂肪组织及纤维组织增多,黏多糖沉积与透明质酸增加,淋巴组织及浆细胞浸润;眼肌纤维增粗,纹理模糊,脂肪增多,肌纤维透明变性、断裂及破坏;肌细胞内粘多糖及透明质酸亦增多伴结膜周围淋巴细胞浸润和水肿。T淋巴细胞仅在眼病的早期起主要作用,但HLA-DR抗原表达发生于瘤性变化的全过程中。因此,早期病变可能以T淋巴细胞作用为主,后期则以成纤维细胞的作用突出而导致纤维组织增生和纤维化。
(三)胫前黏液性水肿为GD的特异体征
光镜下,病变皮肤可见黏蛋白样透明质酸沉积,伴多数带有颗粒的肥大细胞、吞噬细胞和成纤维细胞浸润;电镜下,有大量微纤维伴糖蛋白及酸性GAG沉积,与重度甲减(黏液性水肿)的皮下组织粘多糖浸润的组织学相似。
(四)非特异性病变累及多种组织
久病者或重度甲亢患者肝内可见脂肪浸润、局灶性或弥漫性坏死、萎缩、门脉周围纤维化乃至肝硬化。破骨细胞活性增强,骨吸收多于骨形成,引起骨质疏松。颈部、支气管及纵隔淋巴结和脾脏可增大。
(一)治疗原则
目前对GD治疗方案的选择意见并不一致。在欧洲多优先选用手术治疗,理由是GD的病因复杂,发病机制尚未阐明,有些GD(如G蛋白α亚基突变)对药物的反应差,有时在肿大的甲状腺组织中还可能隐藏肿瘤或其他病变。而在美国,认为放射性碘治疗的疗效可靠,创伤小,疗程短。除少数患者外,多用放射性碘治疗。GD的治疗方案应个体化。放射碘治疗的主要担心是放射线的致癌作用。有研究总结6641例(男性17.5%,女性82.5%)用131I治疗的患者临床资料(1970~1997年),看不出放射性碘的致癌作用。
一般来说,GD可以通过药物、手术或131I等3种方法之一进行有效治疗,3者的适应证没有绝对界线。在实际工作中,选择治疗方法要考虑多种因素:①医师对治疗方法掌握的熟练程度与经验。②患者的年龄、病程长短、病情轻重及甲状腺肿大程度等。年龄较小、病情轻,甲状腺轻至中度肿大者应选择药物治疗。病情较重、病程长、甲状腺中~重度肿大者应采用131I或手术治疗。③甲状腺巨大和结节性甲状腺肿伴甲亢应首先考虑手术治疗。④患者的意愿、文化程度和经济状况也应考虑。⑤药物治疗虽最安全,但疗程长、治愈率较低;如患者较急躁、缺乏耐心,迫切希望迅速治愈甲亢,则应采用131I或手术治疗,不能长期坚持服药者也应采用其他方法。⑥131I治疗快捷、方便、效果可靠,但治疗后甲减的发生率高,对治疗后不能定期随访或不愿接受终身服用TH制剂者不应采用131I治疗。
(二)药物治疗
1.支持治疗/禁碘/戒烟是药物治疗的基础措施
应予适当休息。注意补充足够热量和营养,包括糖、蛋白质和B族维生素等。精神紧张、不安或失眠较重者可给予苯二氮䓬类镇静剂。抗氧化剂、营养支持和心理支持治疗对甲亢患者的恢复有益。在高代谢状态尚未改善以前,患者可采用高蛋白、高热量饮食。除糖类外,可使用牛奶、豆浆、瘦肉、鸡蛋、鱼、肝等食物,在两餐基本饮食之间可加牛奶、豆浆、甜食品。患者出汗多,丢失水分多,应保证足够的饮料,平时不宜喝浓茶、咖啡等刺激性饮料,禁食含碘食盐与食物和戒烟对GD,尤其是TAO的防治有积极意义。
2.抗甲状腺药物控制甲亢
药物疗法应用最广,但仅能获得40%~60%治愈率;其优点是:①疗效较肯定;②不会导致永久性甲减;③方便、经济、使用较安全。其缺点是:①疗程长,一般需1~2年,有时长达数年;②停药后复发率较高,并存在原发性或继发性失败可能;③可伴发肝损害或粒细胞减少症等。
(1)常用药物
常用的抗甲状腺药物分为硫脲类和咪唑类两类。硫脲类有甲硫氧嘧啶(methylthiouracil,MTU)及丙硫氧嘧啶(propylthiouracil,PTU),半衰期1.5小时;咪唑类有甲巯咪唑(thiamazole,MM,他巴唑)和卡比马唑(carbimazole,CMZ,甲亢平),半衰期6小时,所以轻~中度的GD患者每天服药1次即可。两类抗甲状腺药物的结构见图4。硫脲类和咪唑类抗甲状腺药物的作用机制基本相同,都可抑制TH合成,如抑制甲状腺过氧化物酶活性,抑制碘化物形成活性碘,影响酪氨酸残基碘化,抑制单碘酪氨酸碘化为二碘酪氨酸及碘化酪氨酸耦联形成各种碘甲腺原氨酸。近年发现,此组药物可轻度抑制免疫球蛋白生成,使甲状腺中淋巴细胞减少,血TSAb下降。但两类药物亦有一些差别(表6),其中PTU还在外周组织抑制5’-脱碘酶而阻抑T4转换成T3,故首选用于严重病例、甲亢危象、妊娠与哺乳妇女或对MM过敏者;而MM已经成为甲亢的一线用药,尤其适用于儿童GD、肝病患者和对PTU过敏者。此外,未经治疗的GD患者血清脂质过氧化活性增强,血浆巯基(thiol groups)和巯基裂解物水平下降,细胞内抗氧化酶活性增加,而细胞外的自由基清除系统活性不足。MM有助于逆转这些异常。
表6 甲巯咪唑和丙硫氧嘧啶的比较
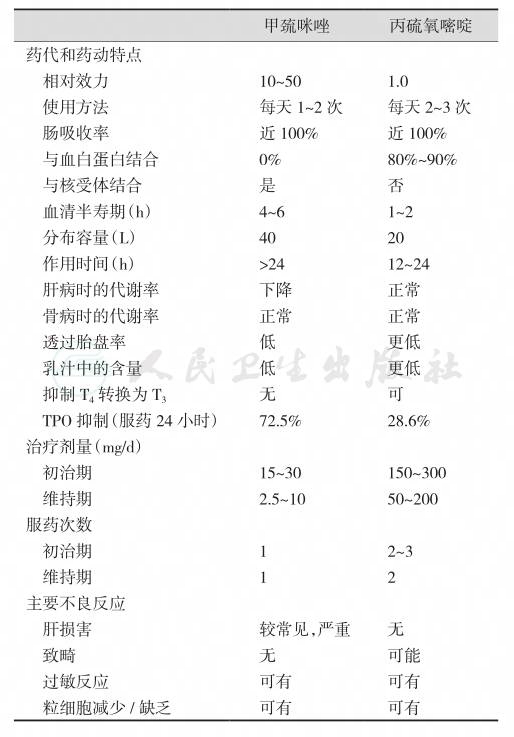
短期用MM治疗可使部分GD的病情得到长期缓解,而另一些患者即使长期使用药物治疗也很难达到这一目的。如果患者的血清IgE正常,易于用MM控制症状。Th淋巴细胞被激活后,合成过多IL-13,后者兴奋B细胞合成TSHR抗体和IgE,因而在用MM治疗时,如TSH封闭性Ig下降不明显,其缓解率低。
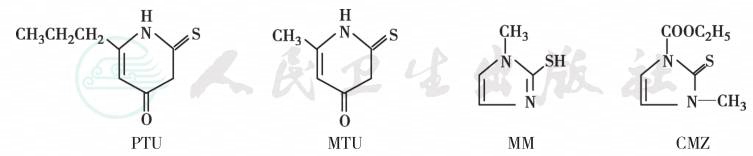
图4 抗甲状腺药物的结构与名称
注:PTU:丙硫氧嘧啶;MTU:甲硫氧嘧啶;MM:甲巯咪唑;CMZ:卡比马唑
(2)剂量与疗程
长程治疗分初治期、减量期及维持期。
1)初治期
MTU或PTU 300~450mg/d,或MM,或CMZ 30~40mg/d,分2~3次口服(初治期和应用大剂量时不主张使用每天单次疗法)。至症状缓解或血TH恢复正常时即可减量。
2)减量期
约每2~4周减量1次,MTU或PTU每次减50~100mg,MM或CMZ每次减5~10mg,待症状完全消除,体征明显好转后再减至最小维持量。
3)维持期
MTU或PTU 50~100mg/d,MM或CMZ 5~10mg/d(如必要可使用每天单次疗法),如此维持1.5~2年。必要时还可在停药前将维持量减半。
疗程中除非有较严重反应,一般不宜中断,并定期随访疗效。治疗中如症状缓解而甲状腺肿或突眼反而恶化时,抗甲状腺药物可酌情减量,有人建议加用L-T4 25~50μg/d或甲状腺粉20~60mg/d,但效果不明。长程(>1年半)治疗对轻、中度患者的治愈率约为60%;短程(<6个月)治疗的治愈率约为40%,但在停药后3个月~1年内易复发。
在GD的药物治疗过程中,一般可观察到血TSH的以下4种变化:①血TSH逐渐升至正常:约占治疗GD患者的50%;血TSH恢复至正常的时间不定,一般为3~6个月;血TSH迅速恢复正常说明药物治愈的可能性大。②血TSH持续降低:部分患者的T3和T4在药物治疗后数个月内达到正常,但血TSH长期≤0.1mU/L,这种情况提示药物治疗的疗效不满意,停药后复发的可能性大。③血TSH波动过大:有些GD患者的血TSH波动在0.1~10mU/L范围内,而临床不伴有甲亢或甲减表现,提示患者对抗甲状腺药物敏感,用药物治愈的可能性大,但需及时调整药物剂量;必要时可加用TH制剂(尤其是儿童患者)。④血TSH正常伴TgAb和(或)467TPOAb抗体明显升高:这种情况往往提示并发自身免疫性甲减的可能性大,不主张采用131I或手术治疗。
(3)不良反应
抗甲状腺药物治疗的常见不良反应是粒细胞减少和药物性甲减,PTU容易发生肝脏或心肌损害;而粒细胞缺乏症与ANCA相关性小血管炎为抗甲状腺药物治疗的严重并发症,且预后较差。
1)粒细胞减少和粒细胞缺乏症
一般为粒细胞减少(MTU多见,MM次之,PTU最少),严重时可致粒细胞缺乏症。前者多发生于用药后2~3个月内,也可见于任何时期。如外周血白细胞低于3×109/L或中性粒细胞低于1.5×109/L,应考虑停药,并应严密观察。GD用MM治疗后可出现贫血,血清中存在MM依赖性抗红细胞抗体,这些抗体可与Rh复合物蛋白结合,但与其他血细胞不结合,有些患者可合并粒细胞减少和血小板减少。有时也出现抗中性粒细胞特异性Fcr受体Ⅲb抗体和内皮细胞-血小板黏附分子-1(plateletendothelial cell adhesion molecule-1,PECAM-1,CD31),而导致中性粒细胞和血小板减少。MM还可引起皮疹、嗜酸性粒细胞增多、血管神经性水肿等。
粒细胞减少的处理原则和方法是:①试用一般性升白细胞药物,如维生素B4、鲨肝醇、利血生、脱氧核糖核酸、碳酸锂等。必要时,每日皮下注射重组的人粒细胞集落刺激因子(rhG-CSF)2~5mg/kg或重组的人粒细胞-巨噬细胞集落刺激因子(rhGM-CSF)3~10μg/kg。白细胞正常后停用;②必要时给予泼尼松30mg/d口服;③粒细胞减少合并药疹较常见,可用抗组胺药物控制,不必停药,但应严密观察,如皮疹加重,应立即停药,以免发生剥脱性皮炎;④粒细胞减少合并中毒性肝炎应立即停药抢救,其治疗方法有糖皮质激素、小剂量TH(甲亢已控制时)、溴隐亭和其他抗组胺药物、红细胞生成素、新鲜全血(少量多次)、骨髓移植。
2)ANCA相关性小血管炎
多见于中青年女性,为PTU的较特异性不良反应。抗中性粒细胞胞浆抗体(antineutrophil cytoplasmic antibody,ANCA)相关小血管炎包括Wegener肉芽肿、显微镜下多血管炎和变应性肉芽肿性血管炎。ANCA以正常人中性粒细胞为底物检测到的自身抗体分为c-ANCA(胞浆型)和p-ANCA(核周型)两种。该抗体对血管炎的诊断极有帮助,尤其是c-ANCA对于Wegener肉芽肿(Wegener granulomatosis)具有较高特异性(98%),而p-ANCA对疾病诊断的特异性相对较差。这些药物偶可诱导产生ANCA血管炎。血清学检查结果与红斑狼疮改变一致,PTU与髓过氧化物酶发生反应形成反应中间产物,而反应中间产物促进自身免疫炎症反应。一般表现为间质性肺炎、肺出血、干咳和呼吸困难;肾血管炎表现为镜下血尿或肉眼血尿、蛋白尿、肾小球炎和肾功能减退(肺-肾血管综合征);少数侵犯多个脏器(肝、脾等),伴有发热、关节肌肉疼痛、皮疹、紫癜,多数于停用后恢复,个别危及生命。临床上呈全身多系统受累表现时应高度怀疑ANCA相关小血管炎可能,根据临床表现(至少存在三个以上的器官损害,如肾脏、肝脏、五官等),结合PR3-ANCA或MPO-ANCA阳性以及组织病理改变多能作出诊断。
少数重症患者需用大剂量糖皮质激素和免疫抑制剂治疗。有条件者在PTU治疗前测定ANCA抗体,并在治疗过程中监测尿常规及ANCA抗体有助于预防ANCA相关性小血管炎的发生和恶化。
3)药物性甲减
多见于抗甲状腺药物用量过大或疗程过长者,但个体对抗甲状腺药物的敏感性差异很大。药物性甲减的最早表现是治疗过程中的甲状腺肿大与血TSH升高。处理的原则是减低抗甲状腺药物用量或暂时停用抗甲状腺药物(但不宜长期停用),在一般情况下,不补充TH(阻滞-替代治疗)。
4)肝功能损害
抗甲状腺药物偶可引起胆汁郁积和肝细胞损害,以PTU多见,而MM一般对肝脏无损害。PTU相关性急性肝衰竭表现为痒疹、黄疸、白陶土样便、尿色加深、关节痛、腹痛腹胀、厌食、恶心或乏力等。30%患者有血清转氨酶升高,4%升高到3倍以上。美国FDA药物不良反
1.Yi BP,Leng SL,Kwang LB,Rootman J. Development of thyroid-related orbitopathy following cataract surgery. Orbit,2009,28(6):383-387.
2.Eckstein AK,Johnson KT,Thanos M,et al. Current insights into the pathogenesis of Graves’ orbitopathy. Horm Metab Res,2009,41(6):456-464.
3.Alkemade A. Central and peripheral effects of thyroid hormone signalling in the control of energy metabolism. J Neuroendocrinol,2010,22(1):56-63.
4.Barreto-Chaves ML,Carrillo-Sepúlveda MA,Carneiro-Ramos MS,et al. The crosstalk between thyroid hormones and the Renin-Angiotensin System. Vascul Pharmacol,2009,52(3-4):102-112.
5.Tribulova N,Knezl V,Shainberg A,et al. Thyroid hormones and cardiac arrhythmias.Vascul Pharmacol,2010,52(3-4):102-112
6.Pothiwala P,Levine SN. Thyrotoxic Periodic Paralysis:A Review. J Intensive Care Med,2010,25(2):71-77.
7.Tsang W,Houlden RL. Amiodarone-induced thyrotoxicosis:a review. Can J Cardiol,2009,25(7):421-424.
8.Costa RM,Dumitrascu OM,Gordon LK. Orbital myositis:diagnosis and management. Curr Allergy Asthma Rep,2009,9(4):316-323.
9.Gonzalez CD,de Sereday M,Sinay I,et al. Endocrine Therapies and QTc Prolongation. Curr Drug Saf,2009,5(1):79-84.
10.Khandwala HM,Van Uum S. Reversible hypercalcemia and hyperparathyroidism associated with lithium therapy:case report and review of literature. Endocr Pract,2006,12(1):54-58.
11.Malm J,F?rnegardh M,Grover GJ,et al. Thyroid hormone antagonists:potential medical applications and structure activity relationships. Curr Med Chem,2009,16(25):3258-3266.
12.Bartalena L,Lai A,Sassi L,et al.Novel treatment modalities for Graves’orbitopathy. Pediatr Endocrinol Rev,2010,7 Suppl 2:210-216.
13.Wilhelm SM,McHenry CR. Total Thyroidectomy Is Superior to Subtotal Thyroidectomy for Management of Graves’ Disease in the United States. World J Surg,2009,34(6):1261-1264.
14.Chong KK,Khanna D,Afifiyan NF,et al. Rituximab Treatment of Patients with Severe,Corticosteroid-Resistant Thyroid-Associated Ophthalmopathy. Ophthalmology,2009,117(1):133-139.e2.
15.Koutras DA. Thyroidopathies. Ann N Y Acad Sci,2000,900:77-88.
16.Earl R,Crowther CA,Middleton P. Interventions for preventing and treating hyperthyroidism in pregnancy. Cochrane Database Syst Rev,2010,(9):CD008633.
17.Karras S,Tzotzas T,Kaltsas T,et al. Pharmacological treatment of hyperthyroidism during lactation:review of the literature and novel data. Pediatr Endocrinol Rev,2010,8(1):25-33.
18.Carrion AF,Czul F,Arosemena LR,et al. Propylthiouracil-induced acute liver failure:role of liver transplantation. Int J Endocrinol,2010,2010:910636.
19.Rivkees SA. Pediatric Graves’ disease:controversies in management. Horm Res Paediatr,2010,74(5):305-311.









