


 收藏
收藏 已收藏
已收藏中文别名 :促甲状腺素( TSH )抵抗综合征
(一)TSH-β亚基/TSHR/Gsα缺陷导致的TSH不敏感综合征
TSH不敏感遵循常染色体显性遗传模式,无不完全外显证据。尽管符合TSH不敏感表型的亚临床和轻度甲低在人群中常见,但遗传因素仍不明了,大部分家系无TSHR缺陷。目前,已经明确的TSH不敏感综合征病因有:①TSH-β亚基基因突变;②TSH受体基因突变;③Gsα亚基基因突变;④TSH受体信号转导途径缺陷(如转录因子PAX8基因突变)。
(二)TSHR功能异常和甲状腺对TSH无反应
TSHR的配体结合区存在一定的特殊性,因为在此区至少存在下列四个特异性结合位点或结构域(图1):①TSH高亲和力结合部位可与TSH受体的拮抗剂或阻滞性TSH受体抗体结合;②TSH特异性结合部位与TSH高亲和力结合部位邻近;③LH/HCG结合部位的特异性不高,仅在血LH/HCG显著升高(如妊娠和胎盘滋养层细胞肿瘤)时起作用;④TSH低亲和力结合部位可与TSH受体激动剂或刺激性TSHR抗体结合。
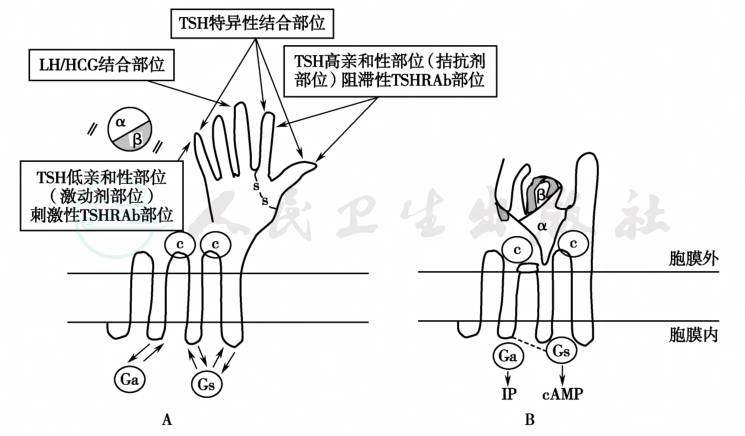
图1 TSHR与配体结合前后的构象变化
A.非激活状态下的TSHR的构象示意图,图中示TSHR的不同结合部位;B.TSHR与TSH或其他配体(如TSHR抗体)结合后,将配体推向穿膜区的胞外环处,并激活G蛋白的信号转导通路,可能是TSH-β亚基与TSHR的胞外结合区(图中的“手”与“手指”)结合,而α亚基与TSHR的穿膜段互相作用,诱发TSHR空间结构变化
1.TSH受体缺陷
目前已发现两种缺陷:一种为TSHR基因突变致TSHR功能异常;另一种为受体后(如cAMP生成)障碍。TSHR基因突变的类型多达20多种[1]。Sunthornthepvarakul等对三例患者及其父母的TSHR基因测序证实,患者的外显子6有两种突变。一个等位基因(T→C)突变导致TSHR的I162N突变;另一个等位基因为C→A突变(P162A)。甲状腺发育不良,血TSH显著升高,T3、T4下降。两种突变型TSHR基因分别由其父母遗传而来,使患者成为复合性杂合子,TSHR有严重功能缺陷。但由于分泌TSH增多,使TSHR缺陷得到代偿,因此可以缺乏甲减的临床表现。Nagashima等报道1对TSH不敏感的日本同胞兄弟,其TSHR基因存在两个新的复合性杂合子突变[2]。血TSH升高,甲状腺位置和甲状腺激素正常,腺体轻度增生。不同基因型一般具有不同的表型,如I167N、P162A和R109Q突变者TSH升高,T3、T4正常;儿童纯合子A553T突变者甲状腺发育不全,伴甲低;受体基因错义突变导致结合TSH减少或完全不能结合,缺乏转导信号。但也有可能具有相同表型,如PAX8和TSHR基因突变患者的甲状腺表型无法区分。无义突变(如W546X)导致受体截短,纯合子无义突变使患者TSH升高,T3、T4降低。天津地区1例先天性甲减患儿在TSHR基因纯合子R450H失活性突变导致该患儿先天性甲减[3]。
2.TSH受体后缺陷
Mediiros-Neto等报道,先天性甲减是由于甲状腺细胞第二信使的cAMP生成障碍,甲状腺球蛋白生成减少。Hager-Malecka等报道的1例42月龄男孩,身材矮小,发育迟滞,伴皮肤干燥、口渴、多尿、隐睾及佝偻病,血 GH、胰岛素、TSH、PTH 等正常或升高,用GH、T3、维生素D等治疗后,血和尿中cAMP无升高,这可能是由于腺苷环化酶系统缺陷而导致了包括TSHR后不敏感综合征在内的多激素(TSH、GH、PTH、胰岛素和儿茶酚胺等)不敏感综合征[4]。Congdon等报道,散发性先天性甲减合并甲状腺增生不良病例存在PAX8转录因子基因的杂合子突变(如A119C和Q40P等),其中1例患者的母亲也有同样突变,而且合并慢性淋巴细胞性甲状腺炎。
3.其他相关性缺陷
主要有:①甲状腺转录因子1(TTF1)基因突变;②TSH基因突变(分子量增大而无生物活性);③Down综合征患儿常伴有先天性甲减;④TSHR阻滞型抗体(TBAb)导致胎儿一过性先天性甲减和甲状腺发育延迟。
此综合征患者的治疗取决于甲减的严重程度。对在临床上无甲减症状,发育正常,血T3、T4正常,只有血TSH增高的患者,一般用L-T4补充/替代治疗,满意的补充/替代治疗的目标是血TSH、T3和T4浓度在正常范围。如果TSHR缺陷严重,出生后又未得到及时治疗,可使患者的身体和智力发育出现严重障碍,以后即使给予甲状腺激素制剂治疗,效果也不满意。甲减时,可用右旋-T4(D-T4)或 3,3,5-三碘甲腺原氨酸(3,3,5 triiodo-thyroacetic acid,RIAC)治疗[7]。









