


 收藏
收藏 已收藏
已收藏生长激素(growth hormone,GH)缺乏症(GH deficiency,GHD)患者的垂体GH分泌量从完全缺乏至正常是一个连续的谱系,因此给GHD下一个明确的定义十分困难。GHD有两个方面的涵义,一是GH量减少而活性正常;二是GH量正常(或升高)但生物活性降低;后者主要见于GH受体(GHR)基因突变(GH不敏感综合征)或原发性IGF-1缺乏症。青春期前发生的GHD又称为垂体性矮小症或垂体性侏儒症(pituitary dwarfism)。
下丘脑-垂体疾病容易发生GHD。按病因,GHD可分为特发性和继发性两类;按病变部位可分为垂体性和下丘脑性两种;按受累激素的多少可分为单一性GHD和包括GH在内的复合型垂体激素缺乏症(combined pituitary hormone deficiency,CPHD)两类。美国的垂体性矮小症发病率为28.7/10万,英国为 2.0/10万,中国北京市的发病率为 11.6/10万(1/8644)。
(一)生长激素基因家族
GH的基因家族分为GH-1、GH-2、绒毛膜促生长素-1(chorionic somatomammotropin,CS-1)、CS-2和CS-P五种。在人类第17号染色体长臂上的60kb区域内,含有 GH-1(GH-N)、GH-2(GH-V)、CS-1(CS-A)、CS-2(CS-B)和CS-P(CS-L)五种GH相关基因,每种基因含5个外显子和4个内含子,长度不超过2kb。垂体GH细胞表达GH-1,其他四种基因由胎盘的滋养层细胞表达。由于GHRH受体基因突变、GH基因突变、GHR基因突变所致的或IGF-1相关的GH不敏感综合征可分为十类。其中GH-1基因的5′-端存在明显的多态性,与GH的分泌及多种疾病有一定关系。此外,垂体不发育症(pituitary aplasia)患者先天性缺乏GH、PRL和TSH,临床表现为低血糖症,GH缺乏的原因未明,其特点是机体发育生长障碍,多伴有骨的畸形或代谢异常。
葛瑞林(ghrelin)是胃分泌的一种肽类激素,含28个氨基酸残基,可与生长激素促分泌物受体(GHS-R)结合,促进GH分泌。实验发现,中枢神经或外周使用葛瑞林可提高食欲,抑制脂肪氧化,导致肥胖,而肥胖患者血中的葛瑞林水平下降。目前认为,葛瑞林是调节生长和能量平衡的一种新的激素,也是联系摄食行为与垂体GH分泌的内分泌调节轴(胃分泌的葛瑞林促进下丘脑-垂体神经肽Y和agouti相关肽的分泌)。此外,葛瑞林还可与中枢神经系统和外周组织的相应受体结合,表现出广泛的生物学作用,如促进PRL和ACTH分泌,对脑-肠-胰轴细胞代谢、心血管功能等均有调节作用,这种GHS的激动剂可望成为治疗多种疾病的有效药物。
胎盘生长激素(placental GH,PGH)为GH-V基因的表达产物,与垂体GH有13个氨基酸残基差异,其促生长作用比GH强而促泌乳作用弱。从妊娠12~20周开始至分娩时,PGH逐渐取代GH的作用,是维持妊娠和胎儿生长发育的必需激素,PGH的分泌受IGF-1和血糖的调节。PGH分泌不足导致胎儿发育迟缓,GH变异和IGF-1相关性GH不敏感综合征分类见表1。
表1 GH和IGF-1相关性GH不敏感综合征分类
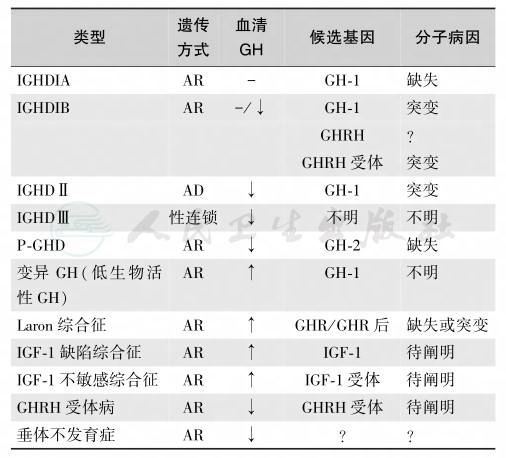
注:IGHD:单一性 GH 缺陷症(isolated GH deficiency);P-GHD:胎盘GH缺陷症(placental GH deficiency);AR:常染色体隐性遗传;AD:常染色体显性遗传;RIA:放射免疫测定;↑:上升;↓:下降
(二)GH的作用
除了人们所熟知的GH作为内分泌激素的作用外,GH还具有促进骨的线性生长、骨重建、骨骼肌生长和免疫调节作用(表2)。儿童至青春期发育成熟的骨骼(长骨)线性生长主要由GH、IGF-1、糖皮质激素和甲状腺激素调节。儿童的躯体线性增长速度是了解有无GH缺乏或GH作用障碍的简单而特异的诊断线索,加用基础血IGF-1测定,或GH兴奋试验可证实诊断。另一方面,药用rhGH可用于多种疾病的治疗。
表2 GH的生理意义和药理作用

一般将循环血液中的信号蛋白分为激素和细胞因子两类,但两种蛋白具有许多相同的理化特征与作用机制。其中一个显著的特点是,无论激素或细胞因子均受细胞因子信号抑制子(SOCS)蛋白的调节。SOCS蛋白家族成员在许多组织中表达,主要起调节生长、骨发育和炎症的作用,但其作用强度不一。慢性炎症性疾病常伴有生长发育障碍,而SOCS蛋白在其中起了重要作用。躯体的线性生长(linear growth)与骨骼发育主要依赖于GH的作用。临床研究显示,慢性炎症所致的生长发育障碍伴有炎症细胞因子的变化。引起GHD的病因包括GH缺乏、IGF-1缺乏和GH不敏感等方面。病变可来自下丘脑GHRH缺乏、垂体病变(如垂体先天缺如、肿瘤、外伤、放射损伤等)、中枢神经系统感染以及遗传异常。GHD的病因见表3,其中原发性GHD最常见,占50%~70%。
表3 GH缺乏的病因

注:GH不敏感是指临床上有IGF-1缺乏表现,但血GH正常或升高,且对外源性GH存在抵抗;GH不敏感综合征:存在GH不敏感,且有生长障碍和其他躯体异常表现;GH部分不敏感是指存在GH不敏感,但无生长障碍和其他躯体异常表现;Galanin:促生长激素神经肽;PACAP:垂体腺苷酸环化酶激活肽;Pit-1:垂体特异性转录因子-1;GH:生长激素;GHRH:生长激素释放激素;IGF:胰岛素样生长因子
循环血液中的GH和PRL属于垂体来源的内分泌激素,但从理论上讲,每个细胞在特定条件下均可表达基因组的所有基因。许多垂体外组织细胞表达GH和PRL基因,以及GH受体和PRL受体;因此合成和分泌的GH和PRL主要在局部起自分泌和旁分泌调节作用,这种基因称为渗漏基因(leaky gene),这种生理现象称为渗漏基因表达现象。除了GH和PRL外,垂体外组织(神经细胞、免疫细胞、生殖细胞和呼吸道细胞)也表达其他垂体激素,主要调节局部组织(尤其是胚胎组织)的生长发育和代谢。在病理情况下,这些旁分泌激素可引起肿瘤或成为肿瘤的一种分泌功能。
(三)遗传性GH缺乏的临床类型
GH基因位于第17号染色体长臂,含5个外显子和4个内含子,前者有2个为GH基因(GH-N,GH-V),3个为绒毛膜生长PRL基因。多数家族性GHD为常染色体隐性遗传,少数为常染色体显性或伴性遗传,可表现为单一性GHD或多发性垂体激素缺乏症或GH作用障碍。许多基因突变可导致GHD(表4)。家族性GHD可根据其遗传方式和基因缺陷种类分为多种类型。
表4 GH缺乏症致病基因
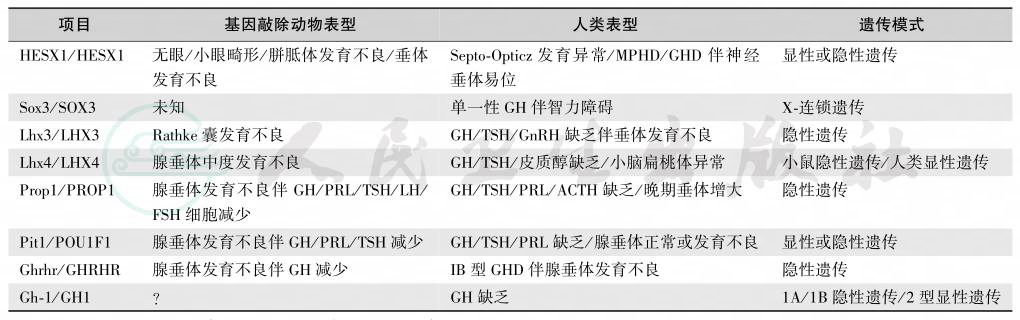
注:MPHD:多种垂体激素缺乏症;IGHD:特发性垂体激素缺乏症;GHD:GH缺乏症
1.遗传性GHD
遗传性GHD分为四类:①遗传性GHDⅠA型:常染色体隐性遗传,ⅠA型是由于GH-1或GH-N基因纯合子突变或缺失,基础GHD或极低,刺激后仍检测不到,是最为严重的GHD。出生后即有生长缺陷,伴低血糖症,部分患者在GH治疗后可检测到高滴度的GH抗体。②遗传性GHDⅠB型:常染色体隐性遗传,GH-N基因正常,但内源性GH减少,GH刺激试验有反应,GH峰值多<7μg/L,外源GH治疗有效,可能是缺乏GHRH或GHRH生物活性降低所致。③遗传性GHDⅡ型:常染色体显性遗传,有明显的低血糖倾向,GH-N基因和GHRH基因均正常,输入GHRH后GH分泌增加,与ⅠB型的区别是遗传方式不同,可能与基因表达受阻有关。④遗传性GHDⅢ型:X-性连锁遗传,同时伴有低γ-球蛋白血症(缺乏 IgG、IgA、IgM 和 IgE),该病是由于 X染色体的两个等位基因(分别负责免疫球蛋白和GH分泌)缺失或突变所致(XLH,OMIM 307200)。
2.家族性全腺垂体功能减退性矮小症
家族性全腺垂体功能减退性矮小症(familiar panhypopituitary dwarfism)有GH不足和其他促激素不足表现。可分为两型,1型为常染色体隐性遗传,2型为性连锁隐性遗传。其他多种激素发生缺乏的先后次序为LH、FSH、TSH和ACTH,同家族中的患者除GHD外,个体所缺乏的其他激素可有不同。GHRH试验示GH释放减少,说明病变可能在下丘脑,但亦不能除外垂体病变。
3.GH抵抗综合征
GH的生成、分泌或生物活性异常可由一种或多种遗传性变异引起,涉及的基因与下丘脑垂体发育、GH、GH受体或葛瑞林的作用有关。GH抵抗综合征大致有三种:①Laron矮小症:常染色体隐性遗传,其特点为血GH正常或增高而IGF-1降低,对外源GH治疗无反应或反应很差。患者肝脏缺乏GH受体,IGF-1生成障碍或细胞膜受体缺陷。②Pygmies矮小症:Pygmies为非洲赤道的矮小家族,患者外观很像垂体GHD,虽然血GH正常,但组织对外源性GH无反应。患者在青春期前生长正常,血IGF-1和同龄儿相近,但青春期时血IGF-1减低,缺乏青春期突发生长,外源性GH不能改善生长,因此至成人时身材矮小。③其他GH抵抗综合征:GH受体后缺陷患者血GH很高且有活性,血IGF-1降低,外源GH无促生长作用,但用重组的人IGF-1治疗有效。GH结合蛋白或GH抗体致循环GH作用抑制。IGF合成缺陷(IGF基因缺陷,肝脏疾病等)或IGF抵抗(包括IGF受体缺陷、IGF受体后缺陷、IGF结合蛋白缺乏等)。
4.GH神经分泌功能异常
有些患儿身高较正常低2个标准差以上,生长速度≤4cm/年,骨龄落后≥2年,GH刺激试验显示GH峰值≥10μg/L,但是测24小时或夜间GH分泌节律可发现峰值降低。此类患者是由于中枢神经-下丘脑-垂体系统某部位有轻度损伤(包括神经递质、GHRH分泌减低或生长抑素增多等),称为GH神经分泌功能异常。用GHRH探针证实许多GHD儿童的病变在下丘脑而不在垂体,GHND患儿用GH治疗有效。
5.复合型垂体激素缺乏症
复合型垂体激素缺乏症有多种垂体激素缺乏表现,一般以GHD伴一种或多种腺垂体激素缺乏为特征。病因为下丘脑-垂体区获得性病变(如外伤或肿瘤)或基因突变。腺垂体特异性转录因子-1(pit-1)及其祖先蛋白-1(Prop-1)异常可导致CPHD。pit-1是垂体细胞生长发育和功能成熟的重要转录因子,与胚胎期腺垂体的发育和相关基因的表达密切相关。pit-1基因突变表现为GH完全缺乏,血清基础PRL不可测出或极低,基础TSH为正常低值、降低或不可测及。Prop-1是一种垂体特异转录因子,只表达于胚胎期垂体(时限性表达)。Prop-1启动胚胎期pit-1基因的起始表达并维持其出生后的持续表达。Prop-1突变导致的CPHD除GH、PRL和 TSH缺乏外,还有 LH、FSH或ACTH缺乏表现。
(四)下丘脑-垂体病变引起的特发性GHD
1.下丘脑微小病变
特发性GHD患儿有围生期病变,包括早产、难产、小胎龄儿、严重窒息、发绀及抽搐。可用GHRH兴奋试验来鉴别IGHD患者颅内损伤的部位,在IGHD中约2/3病变部位在垂体水平之上。
2.垂体自身免疫性病变
研究表明,部分“特发性”GHD患者的血清中存在抗垂体GH细胞抗体,有时抗垂体GH细胞抗体出现在GHD前多年,提示这些患者的病因为垂体自身免疫病变。
(五)颅内病变导致的继发性GHD
1.中颅窝肿瘤
肿瘤压迫下丘脑垂体而发生GHD,较常见的为颅咽管瘤、神经纤维瘤、垂体瘤或神经胶质瘤。
2.头颅创伤/鞍区放疗
严重颅脑创伤是GHD的重要原因,多数伴有其他垂体激素缺乏甚至尿崩症,可在颅脑创伤的急性期、恢复期或在颅脑创伤数月至数年后发病。垂体柄断裂综合征常有永久性GHD,伴血清葛瑞林明显升高。
3.感染/自身免疫/浸润性病变
病毒感染多侵犯下丘脑,很少累及垂体,结核、梅毒、酵母样菌感染及肉芽肿常侵犯鞍区。此外,尚有白血病、含铁血黄素等浸润病变、组织细胞增多症等。其中较常见的是Hand-Schller-Christian综合征。
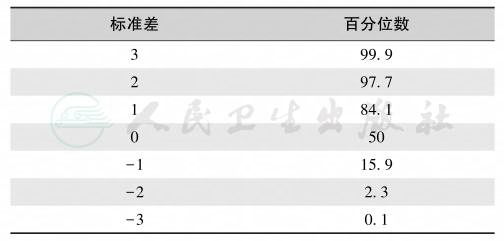
身材异常(身材矮小与身材过高)有同种族同性别同年龄正常人群身高的标准差或百分位数两种表示方法,其对应关系见表6。身材矮小是指身高低于同种族同性别同年龄正常人群身高的2个标准差或第3百分位数的情况。
表6 身材异常的表示方法的对应关系
诊断GHD应强调以下几点:①注重GH的动态变化而非GH的单个点值。②评价IGF-1变化时最好用年龄性别相配的正常参考值,并以0.1百分位数作为临界值;使用高敏IGF-1测定法,判断IGF-1对GH的反应时,GH应≥15μg/L。③筛选试验(运动试验、禁食试验、L-多巴试验和可乐定试验)和确诊试验(胰岛素低血糖试验、精氨酸兴奋试验和胰高血糖素试验)联合应用可提高诊断的特异性和敏感性;一般用禁食试验和可乐定试验作为可疑对象的筛选试验,而将胰岛素低血糖试验和精氨酸兴奋试验作为确诊试验。④胰岛素低血糖试验是公认的“金标准”。⑤实验前空腹,测定的主要指标为血GH、血IGF-1和IGFBP-3;必要时加测垂体的其他激素和胰岛素等。
(一)GHD病例筛查
1.GHD筛查对象
一般从身材矮小和生长缓慢病例中筛查GHD。有些GHD病例的身材矮小或生长速率降低并不明显或仅处于临界水平,此时从早期诊断线索中筛查GHD可提高诊断率,防止漏诊非典型病例。通常患儿6~8岁出现生长迟缓时才被诊断为GHD,而此时已错过治疗的最佳时期,因此早期诊断儿童GHD尤显重要。当患者有以下病史时应考虑GHD可能:①新生儿黄疸期延长、阴茎短小、产时损伤;②头颅放射;③头颅损伤或中枢神经感染;④GHD家族史;⑤颅面中线异常;⑥生长速度缓慢和骨龄延迟;⑦新生儿低血糖症或反复发作的成人低血糖症。
2.GHD筛选方法
GHD的筛选检查方法见表7。
表7 矮小症的筛选检查
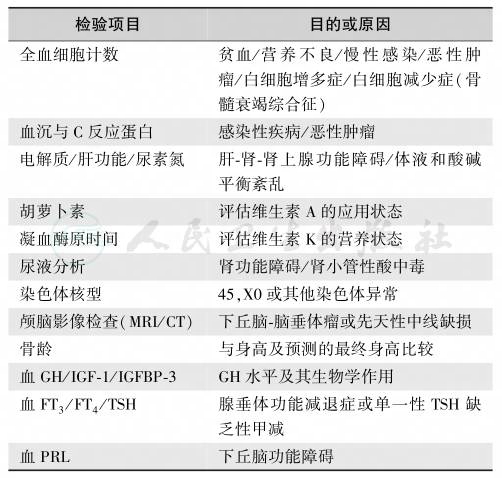
(1)常规筛选检查。①一般检查:如血常规、尿常规及相关生化检查,以了解全身基本情况。一般可根据需要和重点怀疑的病因选择必要的检查,如 T3、T4、FT3、FT4、TSH、ACTH、皮质醇、LH、FSH、PRL、睾酮和雌二醇等。②糖代谢试验:在OGTT中,不少患者在服糖后2小时和3小时血糖偏低。有的可发生低血糖症,严重者因低血糖反复发作而伴有脑损害。③基础GH测定:个体的血GH组分差异很大,用RIA或IRMA法只能测出22kD的GH,仅代表了约25%的血清免疫活性,而且具有抗体依赖性和GHBP依赖性。新一代GH测定是基于GH的生物活性设计的,常用洗脱珠法或免疫功能分析法。由于GH分泌呈脉冲式,峰值与谷值相差较大,故不能仅靠基础GH值诊断本病。④放射学检查:对于生长迟缓超过1年的儿童应常规进行骨龄检查(包括非优势手腕和手掌,<1岁的婴儿检查膝关节和踝关节)。对确诊或怀疑颅内肿瘤、视神经发育不良及中枢神经发育异常的患者还需进行中枢神经MRI或CT检查。MRI需行薄层增强扫描,注意垂体的高度和体积,以及垂体柄和神经垂体的位置。CT虽较MRI分辨率低,但在肿瘤和骨质病变的诊断方面有优势。
(2)血IGF-1测定:可作为GHD的首选指标,对GH缺乏和IGF缺乏的鉴别也有重要意义,但应注意以下几点:①IGFBP干扰测定结果,因而在检测系统中要先用大量IGF-2或放射标记的IGF-1类似物去除IGFBP;②IGF-1水平的变化与年龄有关,5岁以内的血浓度最低;③除GHD外,血IGF-1降低可见于原发性或继发性GH抵抗综合征,如Laron综合征、营养不良或肝脏疾病等;④成年发病型GHD和由脑肿瘤与头颅放射治疗引起的儿童发病型GHD,血IGF-1和IGFBP-3往往正常。
(3)其他筛选检查。①IGF-2测定:IGF-2测定可能有助于提高GH测定和IGF-1/IGF-2测定的一致性,因为正常儿童出现IGF-1和IGF-2同时降低的情况十分少见。如果GH降低,IGF-1和IGF-2均正常,一般不能认为是异常的;相反,如果GH正常而IGF-1和IGF-2降低,或IGF-1正常而IGF-2降低,则多属异常。②IGFBP测定:IGFBP测定对IGF缺乏的诊断和鉴别诊断有意义。IGFBP的血浓度高,受GH的影响明显;但其测定较简单,且受年龄和营养状况的影响较小。
(二)GH动态测定和GH兴奋试验
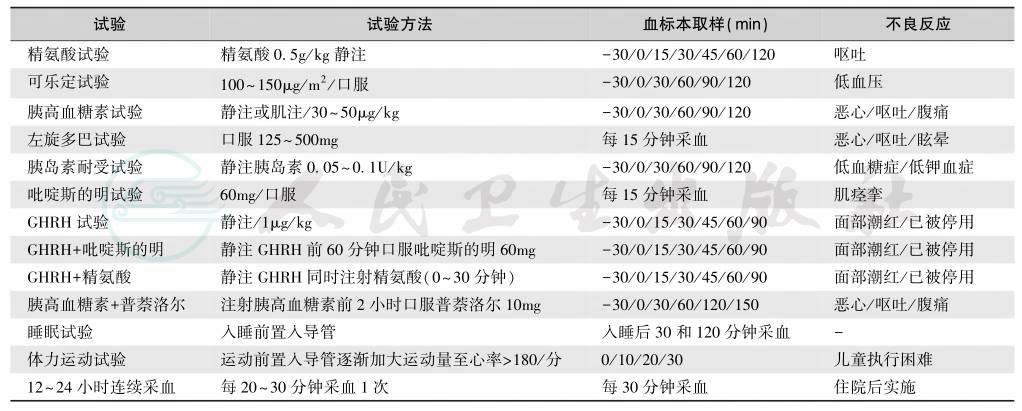
GHD的确诊试验见表8。比较GHRH-ARG试验、胰高糖素试验、精氨酸试验和GH促分泌剂试验的应用经验、敏感性与特异性,目前认为,胰岛素耐受试验仍然是首选方法,其次是胰高血糖素试验。
表8 生长激素兴奋试验
1.动态GH测定
静脉插管,每5~20分钟采血1次,或多次随机采血,共24小时。但GHD与正常人的GH分泌的动态值有重叠。尿GH测定有一定意义,但需用体重和尿肌酐校正。有时儿童GHD的诊断十分困难,因为药理试验并不能完全反映GH的生理分泌状态。例如,运动是GH分泌的最强刺激,但运动试验则因运动量和个体(尤其是肥胖者)的反应性差异而有不同的表现。有时,肥胖/超重者的基础GH分泌可能低至GHD水平,但血IGF-1正常(游离IGF-1可能升高);体重恢复正常后,GH分泌亦随之转为正常。因此,在分析运动试验结果时,应该特别注意体重的影响,肥胖、超重和体重减轻者应该有不同的GHD诊断值,避免误诊为GHD。
2.GH兴奋试验
生长激素兴奋试验很多,单一的兴奋试验只针对GH分泌的某一途径而不能探知其复杂的生理调节过程。各种GH兴奋试验的共同缺点是:①正常值的个体差异大,如将诊断GHD的血清GH分割点定为10μg/L,患者和正常人的重叠率可达30%;②测定GH的方法各异,正常值的表示方法(mU/L或μg/L)也不同,由于使用的GH抗体与GH的亲和性不同而产生较大误差;例如,免疫功能分析测得的GH值要比免疫放射分析(IRMA)法低1~2倍。如果使用IFA法,其分辨值应重新确定;③年龄、青春期发育、体重、应激与代谢影响GH分泌,有的儿童在使用性腺类固醇激素后,GH兴奋试验转为正常,因而青春期发育前患者应在必要时使用性腺类固醇激素“点火”,大约可使一半的GHD患者转为正常反应。单一性药理GH兴奋试验的缺点是:①试验为非生理性的,每一种刺激物都有各自特定的GH分泌机制,但却无法反映GH分泌的整体生理状态;②药理试验存在不良反应;③首选混合性药理GH兴奋试验,可能较单一性药理试验更合适,因为减少了GH分泌反应的变异性。
一般选择可乐定试验、精氨酸兴奋试验、胰岛素低血糖试验和胰高血糖素试验中的两种做GH兴奋试验。根据结果进行如下决策:①如果兴奋试验的所有GH均低于10.0μg/L,应进行进一步检查(头部MRI、CRH兴奋试验,或GH基因、GHR基因突变分析等);②如果 GH峰值低于15μg/L,应测定 GHBP;当 GHBP降低时,应做 IGF生成试验;异常者行 IGF-1治疗;③如果 GH峰值高于15μg/L而GHBP正常,继续临床观察;④如果GH峰值为10~15μg/L,应在6个月内复查。
(1)口服蛋白餐试验:
蛋白餐(protein meal)可引起更为广泛而强烈的生理性GH分泌,因而是一种高效而相对敏感的GH分泌兴奋试验。生理性GH兴奋试验主要包括睡眠试验、运动试验和24小时动态监测,但应用欠方便。新近提出了营养试验,用豆蛋白(精氨酸和赖氨酸的含量高,比值约为1∶1)0.6g/kg,刺激 GH的分泌强度等同于运动试验和睡眠试验,具有一定的应用价值。本试验无不良反应,饮食蛋白可通过多种途径和机制刺激GH分泌,并可兴奋胃肠葛瑞林分泌及胰岛素释放。
(2)胰岛素低血糖-GH刺激试验:
胰岛素低血糖-GH刺激试验是目前推荐的常用试验。低血糖刺激脑内葡萄糖受体,激活单胺类神经元通过α2受体而促进GHRH分泌,同时抑制生长抑素分泌。用普通胰岛素0.1U/kg加入2ml生理盐水中一次静脉注射。试验前及试验后30、60和90分钟采血测GH和血糖。血糖<2.8mmol/L或比注射前血糖值降低50%以上为有效刺激;体质较健康及原有低血糖症(如慢性肝病、糖原累积病)者应使血糖降至2.2mmol/L.如刺激后GH峰值10μg/L以上为正常反应,<5μg/L为反应低下,3~5μg/L为可疑,<3μg/L可确诊为 GHD。
(3)胰高血糖素试验:
用于评价成年GH的储存功能。患者午夜后禁食,次日上午8~9时卧位状态下,静脉注射胰高血糖素1mg(溶解于10ml生理盐水中),分别于 0、30、60、90、120、150、180、210和240分钟采血测定 GH 和血糖。 正常人大约于90分钟时GH升高,以后逐渐下降(不作为诊断依据);GH升高至3μg/L;低于此值提示GH分泌不足或缺乏。
(4)L-多巴-GH刺激试验:
L-多巴通过刺激GHRH促进GH的分泌。患者餐后服L-多巴制剂500mg(体重15~30kg者口服250mg)。服药前及服药后30、60、90和120分钟分别采血测GH值。正常人60~120分钟时GH≥7μg/L,垂体性矮小者无反应。于口服L-多巴前20分钟内运动(如上下楼梯20次左右)可提高试验的反应性(运动-L-多巴试验)。
(5)葛瑞林刺激试验:
葛瑞林为内源性GH释放肽受体的配体,可强烈刺激GH的分泌。但方法学仍未统一。
(6)GHRH和精氨酸试验:
数年前,人们广泛应用GHRH加精氨酸(GHRH-ARG)试验来代替胰岛素耐受试验。但是,2008年重组的人GHRH被停用。
3.GH动态试验结果分析
目前的GH兴奋试验存在许多问题,应特别注意以下几点:①GH兴奋试验为非生理性试验,而所有的药理试验都不能观察到GH的生理性分泌特征,所以分析结果时要结合临床资料,并尽可能进行动态追踪观察,才能获得正确诊断。②GH兴奋试验的结果分析无统一的标准,根据GH兴奋试验结果判断为“GH分泌储备能力降低”只是一种主观臆断。最早的GH判定值为2.5μg/L,后来增至7.0μg/L;而应用重组的人GH后,又进一步增至10.0μg/L;但对GH判定值调整仍缺乏有力依据。③血GH水平对年龄和性腺类固醇激素具有强烈依赖性。青春期发育前的GHD诊断要特别慎重,许多“GH分泌储备能力降低”的儿童在青春期发育时或在应用性腺类固醇激素诱导青春期发育后,其GH分泌和生长发育可完全恢复正常。④GH的测定技术有限。目前的GH测定的敏感性仍不理想,重复性较差。一方面,GH的测定误差最高可达3倍;另一方面,测定GH的下限值不甚敏感。新近发展起来的“免疫功能性GH”测定只测定能与GHBP结合的GH,故其结果与临床的符合程度明显提高,但仍有50%的正常儿童在GH兴奋试验后,其GH峰值低于7.0μg/L。⑤GH兴奋试验存在不少不良反应,有时还可发生严重低血糖反应或肝损害。
(三)GHD诊断
GHD的诊断尚无统一的标准,确立GHD诊断的依据也是相对性的。评价GHD的体检指标应包括身高、身体的上部量和下部量、骨龄以及生长速度等;实验室指标应包括GH、GHBP、IGF-1。如果儿童的生长速度正常,GHD和IGF-1缺乏的可能性极小;当没有发现垂体GH分泌异常,一般不必做GH动态试验。怀疑为IGF-1缺乏时,还应包括IGFBP。如果血IGF-1降低伴IGFBP改变提示为GH分泌或GH活性异常,应对下丘脑-垂体-IGF-1系统进行全面检查。
1.国际GH协会诊断标准 主要是:①身高低于同年龄、同性别正常人2个标准差,并排除Turner综合征、甲减、慢性系统疾病等其他影响生长发育的疾病。②基因诊断(如Prop-1和POU1F1突变)。③骨龄检查、中枢神经系统MRI和CT检查发现病变。④GH激发试验的血GH峰值<10μg/L,伴或不伴血IGF-1及IGFBP-3降低(低于同年龄、同性别正常人2个标准差)。
2.上海市儿科研究所诊断标准 主要是:①身高低于同年龄、同性别正常人2个标准差或第3百分位(根据Stadiometer测定)。②生长速率<4cm/年。③骨龄落后于同年龄、同性别正常均值 2年以上(根据 Greulich-pyle图谱评价)。④三种GH激发试验(L-多巴、可乐定及GHRH)的血GH峰值均<10μg/L。⑤排除引起生长迟滞的其他疾病。
3.湘雅代谢内分泌研究所诊断标准 主要是:①身高增长率<4cm/年,较同年龄同性别正常人均值低2个标准差以上。②典型临床表现,面容体态幼稚,第二性征发育延迟或缺乏。③骨龄检查结果均较实际年龄落后2年以上。④L-多巴及胰岛素低血糖激发试验示GH峰值<5μg/L。⑤排除体质性身材矮小、器质性疾病、内分泌代谢疾病及遗传病。
4.成人GHD诊断标准 应用两种GH兴奋试验,其中以胰岛素低血糖试验最为可靠。血GH、IGF-I及IGFBP-3降低仅提示GHD可能,不能代替GH兴奋试验。必要时应作其他垂体和靶腺激素测定以协助诊断。但是,如果患者已有两种以上的垂体激素缺乏的依据,则没有必要再做GH兴奋试验。
5.肥胖儿童GHD诊断标准 GH缺乏儿童常伴有矮小和肥胖,而胖者的自发性与刺激后GH分泌减弱。Stanley等发现,在116例2~18岁的儿童中,BMI的标准差积分(BMI SD score,BMI SDS)与GH刺激试验GH峰值的自然对数呈负相关,而身高的标准差积分(height SDS)与IGF-I、IGF-结合蛋白-3、年龄、性别及青春期发育无相关,经校正后,BMI标准差积分与GH刺激试验GH峰值的自然对数仍呈负相关。以GH峰值10、7和5μg/L作为切割值时,BMI SDS均是 GHD诊断的独立影响因素。因此,肥胖儿童容易出现GHD的过度诊断问题。
6.青春发育期的GH分泌试验与GH缺乏症再评价由于相关基因突变引起的多种垂体激素缺乏者的GH缺乏为永久性的,但其他原因所致的GH缺乏者的GH分泌反应在青春发育的后期可能转为正常,其自动恢复的原因见表9。
表9 GH刺激反应转为正常的原因

儿童发病的GH缺乏症(CO-GHD)在青春发育期进行再次试验时,常用生长速度和骨龄评价药物治疗疗效。青春转型期GH缺乏症的再评价与诊断流程具有许多特殊性,此时为了避免假阳性,应预备1~3个月的GH洗脱期,并推荐采用胰岛素低血糖兴奋试验、GHRH兴奋试验和精氨酸兴奋试验,见图1。
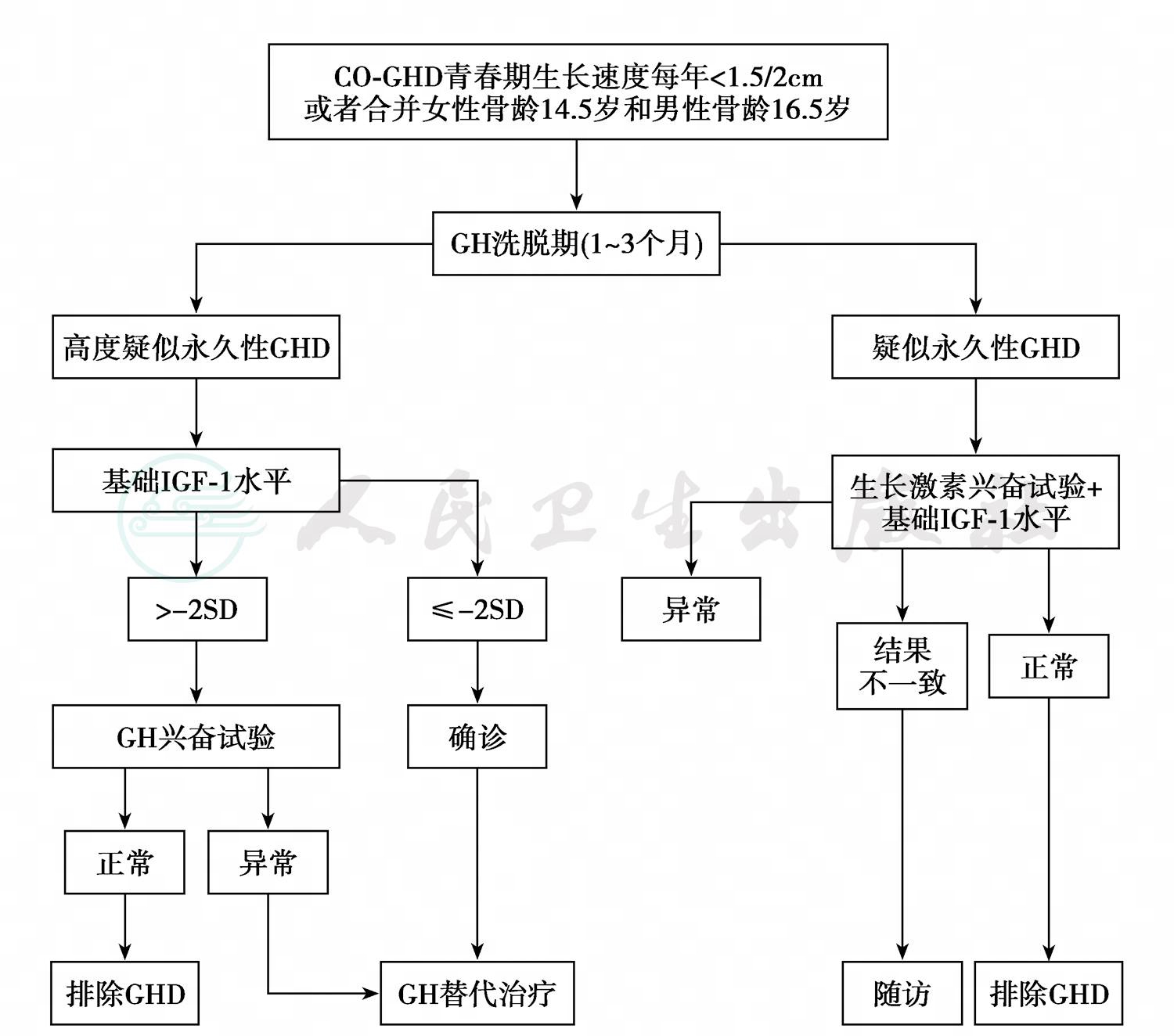
图1 青春转型期GH缺乏症再评价与诊断流程
(四)GH抵抗的诊断
GH抵抗者测定GH生物活性/血IGF-1/IGF-1受体功能。当血IGF-1、IGF-2和IGFBP-3下降而GH升高时,要想到GH不敏感综合征的可能。GHR缺陷(突变)常有家族史(常染色体隐性遗传),Laron矮小症患者的血清GH免疫活性测定正常或升高,但IGF-1低下(由于GH受体缺陷)、GH抵抗综合征患者血清GH正常或增高,受体结合活性明显降低,血清IGF-1低于正常,但应用外源性GH治疗后生长加速。先天性IGF-1抵抗患者的血清GH基础值及兴奋试验均为正常反应,IGF-1升高,IGF-1活性正常,但存在IGF-1受体或受体后缺陷。Pygmies矮小症患者血清GH正常(或增高),IGF-1降低,IGF-2正常,外源性GH不能改善生长状况。当血IGF-1、IGF-2和IGFBP-3降低,而血GH升高时,提示GH受体突变所致身材矮小症,如有身材矮小家族史,则进一步支持诊断。GH不敏感综合征的诊断依据是:①血清GH基础值≥10U/L(约5μg/L);②血IGF-1≤50μg/L;③身高低于同年龄、同性别正常人的第3百分位数;④血GHBP≤10%(125I结合率);⑤GH治疗后,血IGF-1升高≤2倍,IGF-1和IGFBP-3对GH刺激的反应性下降是诊断标准中最重要的项目;⑥相关基因突变。
(五)IGF-1生成试验
GH抵抗是引起儿童身材矮小的主要原因。评价GH的反应性和IGF-1生成试验是诊断和鉴别GH抵抗与继发性IGF-1缺乏的重要依据。IGFGT的实施方法很多,其中应用较多的方法是所谓的标准IGF-1生成试验。试验时间为5天,4天注射 rhGH(每天 33μg/kg,总量 132μg/kg)。 诊断切割值定为批内变异系数的2倍或2倍以上,或IGF-1增加绝对值高于15ng/ml。同时测量IGFBP-3可提高敏感性,其切割值为升高0.4mg/L。诊断严重GH抵抗综合征的计分系统见表10。
表10 严重GH抵抗综合征的计分系统

注:GHBP:GH结合蛋白;GHIS:GH抵抗综合征;IGF:胰岛素样生长因子;IGFBP3:IGF结合蛋白3;IGFGT:IGF生成试验
未经治疗的GHD至成年后遗留永久性身材矮小。完全性GHD、生长速度慢的部分性GHD和慢性肾衰竭所致的生长障碍对GH的疗效较好。骨骺已融合者用GH治疗无效,滥用GH制剂还可能导致糖尿病、多毛、股骨头滑脱或肿瘤复发。继发性GHD由颅中窝瘤、颅咽管瘤、垂体瘤或颅内感染与肉芽肿病变引起,其预后不佳。由于躯体疾病引起的身材矮小必须针对原发病进行病因治疗,不能盲目使用GH制剂。
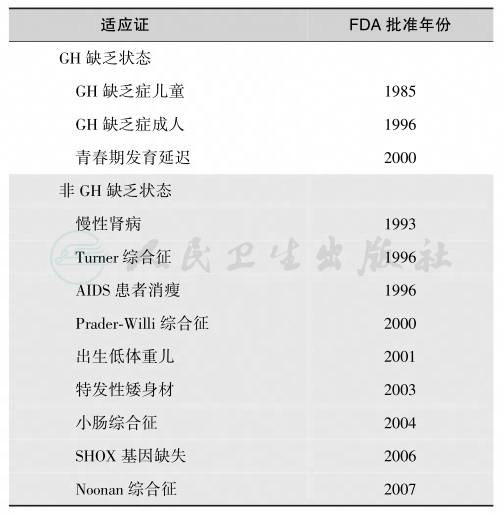
目前,临床应用的rhGH和人GH结构完全相同,rhGH的成功应用使GHD儿童能够基本达到正常身高。儿童GH缺乏性身材矮小的治疗目的是使患者尽量保持正常身高。确诊为GHD者需给予重组的人GH(rhGH)治疗。美国FDA批准的rhGH适应证是:①GHD患儿(特发性矮身材的对象是非GH缺乏的不明原因身材矮小,或身高低于同性别、同年龄儿正常参比值的2.25 SD以上,或预计其成年身高在-2SDS以下者);②AIDS相关的代谢病和消瘦;③Prader-Willi综合征;④宫内发育迟缓患儿出生后持续矮小者;⑤成人GHD替代治疗;⑥Turner综合征、生长障碍与特发性矮小症、SHOX基因突变不伴GHD患儿和Noonan综合征;⑦短肠综合征;⑧儿童肾移植前肾衰相关性生长障碍。
(一)运动和锻炼
运动和锻炼应作为GHD治疗的基础措施和GH补充的辅助治疗。GH和IGF-1具有较强的合成代谢作用,而运动和体育锻炼可促进GH和IGF-1的合成与分泌,并能维持至运动后的一段时间及每天的GH分泌率,GH补充/替代治疗又可增强患者的运动和体育锻炼耐力,提高最大摄氧量、通气阈值、脂肪酸可用性和骨量。因此,运动和体育锻炼应作为GHD的基础治疗和GH补充/替代的辅助治疗。此外,应给予足够的优质蛋白质,因为蛋白餐能有效刺激GH的分泌。
(二)rhGH治疗
对GHD最理想的治疗是用GH补充/替代治疗(表12)。早期应用可使生长发育恢复正常。但是,影响GH治疗疗效的因素很多,如GH的剂量、疗程、开始治疗时的身高、生长速度、年龄等。但是,所有这些因素都只能部分解释患者对GH的个体差异,更重要的因素可能来自GH受体(GHR)本身,即GHR的多态性。GHR的多态性来自GHR基因的外显子3、外显子6和外显子10。其中外显子3多态性是决定增高反应的关键因素。由于在一般情况下,GHR的多态性是未知的,故很难事先预期治疗个体对GH的反应。
表12 GH治疗适应证
1985年,美国FDA首先批准应用rhGH。第一代rhGH由大肠杆菌合成蛋氨酸GH,它是在人的GH的氨基末端附有一额外的蛋氨酸残基,在一定程度上影响了GH三级结构,且有抗原性。第二代rhGH属于不含蛋氨酸的GH单体,其一级和二级结构均与天然GH(22kD)相同,较蛋氨酸GH的抗原性低,但提纯步骤复杂,价格昂贵。第三代rhGH采用分泌型技术,产生的rhGH结构与天然GH完全相同。rhGH治疗可根据美国内分泌学会的GH治疗指南进行。临床应用rhGH必须有明确的适应证。rhGH不能滥用于青少年增高和竞技目的,滥用不但不能获得期望的疗效,有时还会带来严重后果。如果一时不能确定病因,可试用GH或IGF-1治疗1个疗程,无效者应停用。
1.rhGH治疗入选标准
我国rhGH治疗IGHD患者的入选标准是:①身高低于同年龄同性别正常儿童2个标准差以上;②生长速率每年<4cm;③GH药物激发试验(两次)的血清GH峰值<10μg/L,其中GH峰值<5μg/L为完全缺乏,GH峰值>5μg/L为部分缺乏;④骨龄延迟>2年;⑤患者处于青春发育前期(TannerⅠ期);⑥体检未发现先天性遗传代谢性疾病及染色体畸变;⑦药物治疗前血清T3、T4及TSH正常,肝、肾功能及血、尿常规正常。
临床上,可获得较好疗效的治疗对象是:①完全性GHD者至少两项GH兴奋试验的GH峰值≤5~7μg/L。②部分性GHD者生长速度慢,兴奋试验中血GH峰值在5~10μg/L或7~10μg/L。但需注意,有些属正常身材矮小儿童的兴奋试验结果可能也在此范围内。③有头颅放射治疗或中枢神经系统受损病史者虽兴奋后的血GH峰正常,但夜间GH分泌谱低于正常。④慢性肾衰竭所致的生长障碍。⑤患有Crohn病、青少年特发性关节炎、囊性纤维化和营养不良症的儿童如伴有生长障碍可试用治疗,治疗的最佳时间窗是青春期前或青春发育的早期。
此外,FDA还批准用rhGH治疗Turner综合征、Noonan综合征、Prader-Willi综合征、慢性肾衰、矮小同源框基因缺陷症(short stature homeobox-containing gene deficiency,SHOX-D)、先天性低体重儿(SGA)、儿童慢性肾病和特发性矮小症。GH制剂禁止使用于其他原因所致的矮小症(如遗传性矮小综合征、软骨发育不良综合征、呆小病、心因性矮小症等)或意欲增高者。
2.治疗剂量和方法
rhGH的治疗与其他激素补充/替代治疗有所不同,不是随年龄而增加剂量,相反,一般都是随年龄而减少剂量。按完全性GH缺乏的补充/替代量计算,其有效剂量规律是:儿童>成年女性>成年男性>老年女性>老年男性。原来,rhGH的治疗剂量按GH产生率计算得出,但实际上多按临床经验决定。该药的常规剂量为每天25~50μg/kg,在此剂量范围内,增加剂量会提高生长反应,但两者不呈线性关系,剂量增倍,生长反应只增加1/3,故应根据价格-效果方程式,按体表面积或体重计算rhGH剂量。对已进入青春期者,建议剂量偏大,可用至每周1.0~1.3U/kg,分6~7次于睡前皮下注射,目的是仿效正常青春期的高GH分泌方式,克服青春中后期的GH抵抗,获得如同正常人的青春期身高突增。成人型GHD主要为GH补充/替代治疗。国际GH协会推荐初始计量为0.3mg/d(无须考虑体重),美国FDA推荐用量为每天6μg/kg,最大剂量不超过每天25μg/kg(35岁以上者每天不超过 12.5μg/kg),见表13。多数人认为,每日给药比每周注射2~3次的疗效提高25%。间歇治疗(治疗6个月,停药3~6个月)的效果不如连续治疗。临睡前注射使血GH于睡后升高,而采用夜晚注射具有更佳的效果。
表13 不同疾病和不同地区推荐的GH治疗剂量
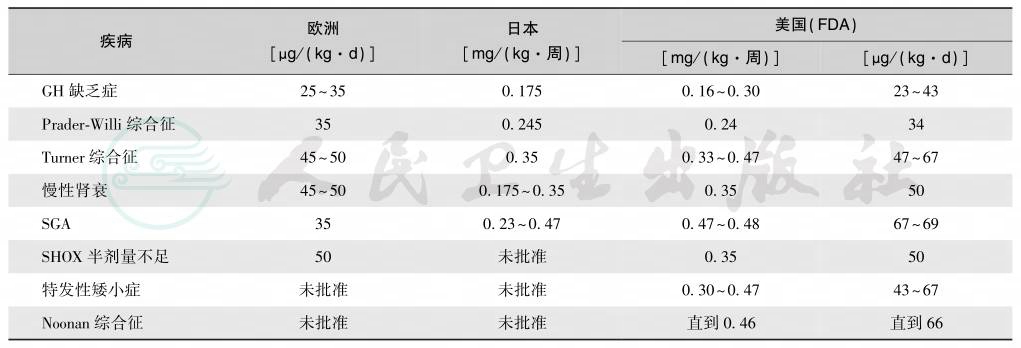
注:SHOX:X染色体矮身材同源框基因;SG:低体重儿
既往用肌内注射避免GH抗体形成,但皮下注射同样是安全有效的。在注射rhGH后,血GH比自发性GH分泌者高,高水平GH的持续时间比正常脉冲长。GH的促生长作用是通过产生IGF-1来实现的,但由于IGF-1主要是旁分泌激素,因此很难由血IGF-1来判断rhGH的长期治疗效果。
3.疗效观察与分析
儿童GH缺乏症治疗有效时,其反应较明显。除了生长速度加快外,应注意体成分、血清IGF-1、BMD、血脂谱和肌肉的变化,同时需要监测糖代谢异常的发生,见表14。
表14 儿童发病的GH缺乏症治疗反应
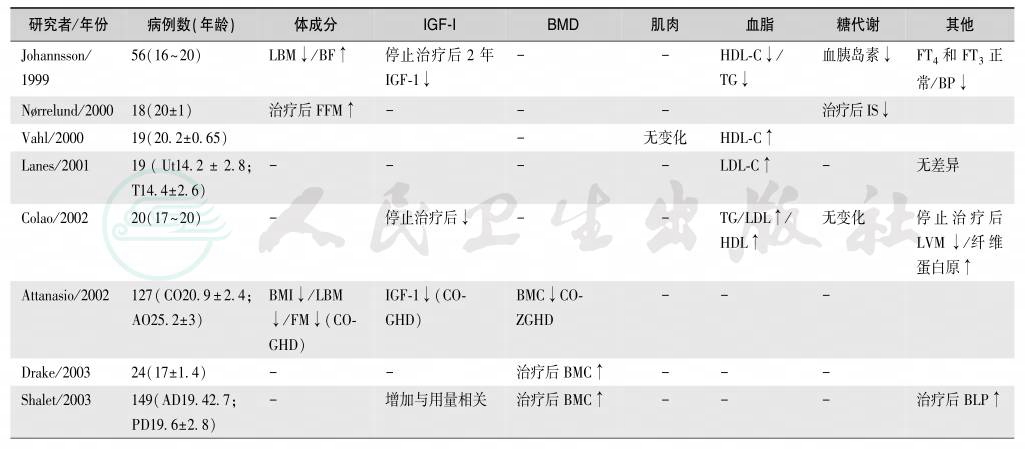
续表
注:BLP:骨源性碱性磷酸酶;LBM:瘦体重;BF:体脂;TG:甘油三酯;SBP:收缩压;TBF:体脂总量;FFM:无脂肪体质;IS:胰岛素敏感性;WC:腰围;LVM:左室质量;BMI:体质指数;BMC:骨矿含量;BMD:骨矿密度;QoL:生活质量;Ut:非治疗组;T:治疗组;CO:childhood-onset GHD,儿童发病的GH缺乏症;AO:adult onset GHD,成年发病的GH缺乏症;AD:成人剂量;PD:儿童剂量;NON-IGHD:非特发性GH缺乏症
(1)身高和生长速度:
第1年生长速度可达8~15cm,第2年仍稍高于正常生长速度,但第3~4年则降到正常的生长速度。有人总结194例 GHD患者用重组人生长激素(Genotropin,健豪宁)治疗1年的效果,青春前期患儿的生长速度由(3.3±1.4)cm/年升至(9.3±2.6)cm/年,青春期患者则由(4.0±1.2)cm/年升至(8.4±1.4)cm/年。 北京协和医院等用rhGH治疗特发性GH缺乏性矮小症患儿1年,生长速度由(2.8±1.0)cm/年增至(13.1±2.5)cm/年,骨龄无明显增加,rhGH用量为每周0.5~0.7U/kg,分6~7次注射。用 rh-GH治疗的GHD患儿,身高、生长速度和血IGF-1是评价生长反应的主要指标;但测定IGFBP-3因无年龄因素的影响而更适合于年幼儿童,优于血IGF-1。血瘦素在治疗后显著降低,开始rhGH治疗时的瘦素水平以及治疗后的变化能反映长期生长反应。
(2)肌力和肌容量:
尚无肯定结论。一些资料显示,长期用rhGH治疗可增加肌力和肌容量,但短期治疗该作用不明显。
(3)其他因素:
除rhGH的剂量和注射频率外,影响疗效的因素还有:①治疗前身材越矮小及生长速度越慢者,生长反应越好;②治疗的第1年由于有追赶生长,生长速度增加最明显,持续治疗后生长速度逐年减慢;③rhGH治疗不仅要考虑所达到的身高,而且应持续到形成峰值骨量之后才能停药;④继发性GHD比原发性GHD的治疗效果好,这是因为继发性GHD患者在患病前已正常生长数年,丢失的生长潜力较少。
4.不良反应
注射rhGH的局部及全身不良反应较少,但应注意rhGH治疗的下列潜在危险性:①胰岛素抵抗:短期使用后,胰岛素敏感性降低,但长期应用数年后,胰岛素的敏感性反而恢复至治疗前水平;②可能使已有糖尿病倾向的患者演变为2型糖尿病或使亚临床型甲减变为临床型甲减,但证据不足。肥胖2型糖尿病使用GH对糖尿病病情可能有不良影响,但需要进一步观察确定;儿童GHD患者伴有2型糖尿病时可以继续使用;③心脏不良反应:如果GHD患者未接受GH治疗,心血管病风险增加,而接受GH治疗者的心血管病风险降低;一般患者使用GH后可改善心肌收缩和扩张功能,但能增加老年患者的左心室质量;④肿瘤风险:下丘脑-垂体肿瘤的风险不增加,但建议治疗前和治疗过程中注意监测;在恶性肿瘤未治愈前,禁止使用GH治疗。患有恶性肿瘤的儿童使用GH可能增加肿瘤复发风险,但无证据支持GH治疗增加成年患者肿瘤发病或复发的风险;⑤其他不良反应:可能引起头骨骺脱位或股骨头滑脱而致跛行或髋部及膝部疼痛,产生GH抗体。此外,头颅放疗常引起GH缺乏症,而给予外源性GH治疗可能增加中枢神经系统肿瘤风险。
GH治疗反应的差别很大,如果治疗中有一定追赶生长反应,但未达到理想的要求程度称为GH治疗反应不良或治疗反应不满意。治疗中应定期评价反应,早期确定治疗反应不满意者,调整治疗方案,取得满意治疗效果(图3)。
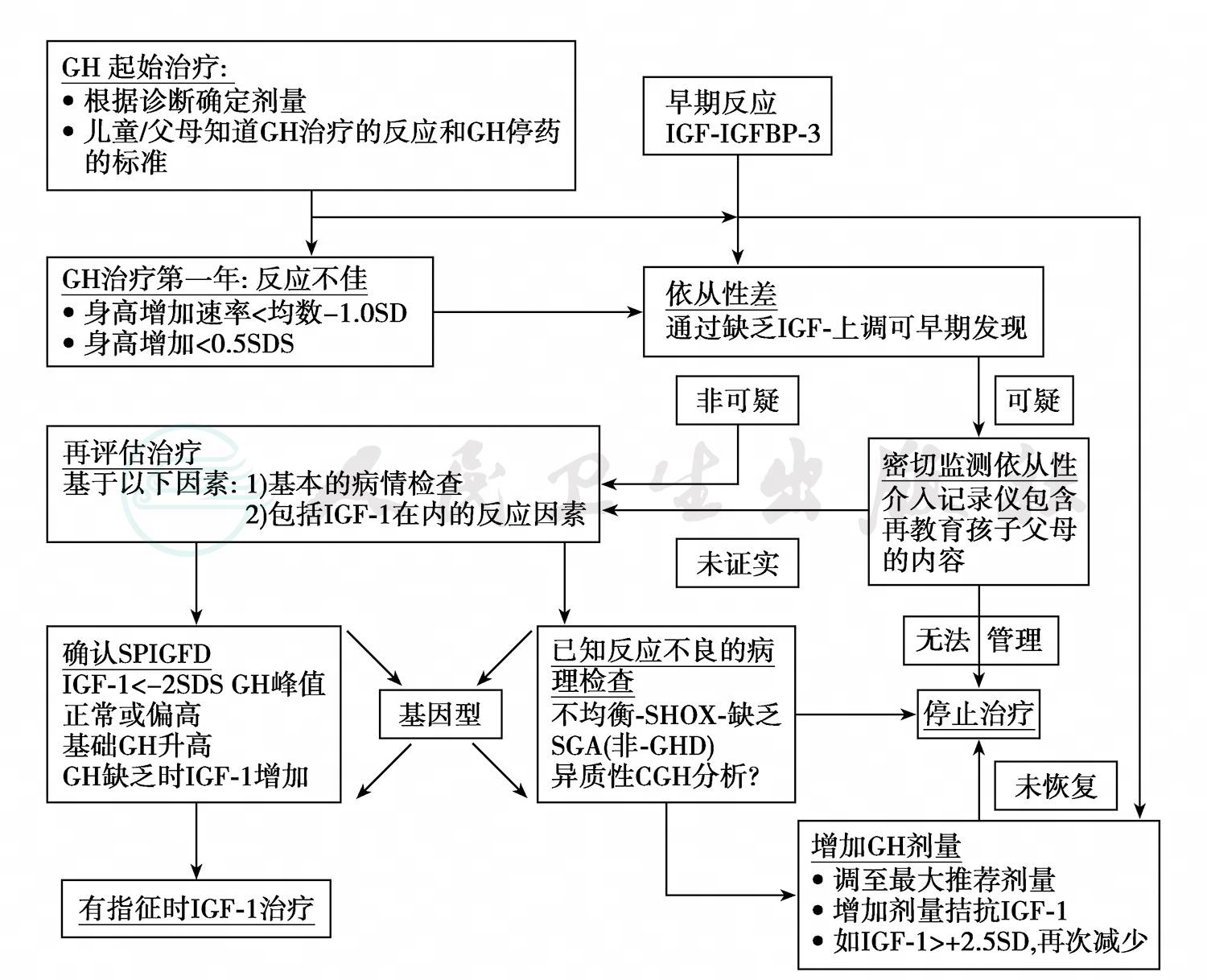
图3 GH治疗反应不佳的处理流程
增高速度低于平均值的-1.0SD,即相当于特发性重症GH缺乏症身高增值标准差分数(SDS)0.4或其他疾病 SDS 0.3;CGH array:比较基因组杂交阵列;SHOX deficiency:矮身材同源框缺乏症;SDS:标准差分数;SGA:小于孕龄儿;SPIGFD:重症原发性IGF-1缺乏症
5.rhGH治疗失败
垂体功能减退患者出现生长障碍的原因不单是GH缺乏,性腺功能减退是重要的原因。对已确诊为GHD者应进行GH补充治疗,如果为GH不敏感综合征,则给予IGF-1治疗;治疗中定期观察病情变化。在rhGH治疗过程中,GH的促生长作用将随疗程的延长而逐渐下降,但只要继续治疗,其作用至少能达到正常的生长速度,此种情况不可视为GH治疗失败。临床上,如rhGH应用不当,难以取得应有疗效,常见于以下情况:①治疗过晚,骨骺已经融合或接近融合;②用量过小或治疗未按要求进行;③用rhGH治疗后迅速产生抗GH抗体;④rhGH疗程太短(<6个月)或虽然接受了长期治疗,但因药品或依从性原因而经常中断治疗;⑤诊断错误,如GH抵抗性矮小症、原发性IGF-1缺乏症、软骨发育不全、营养不良、呆小症(克汀病)、脑中线-视神经发育不良症等;⑥合并多种垂体激素缺乏或躯体性疾病、脊柱放疗等;⑦同时伴有亚临床甲减或垂体激素缺乏症,或补充的糖皮质激素过量。
6.青春发育期GH缺乏症
临床表现是:①线性生长缺乏或减慢;②瘦体重减少而内在脂肪增加;③BMD降低;④IGF-1降低;⑤肌力下降;⑥血脂谱异常(胆固醇,HDLD-胆固醇和甘油三酯升高 L-胆固醇降低);⑦心肌功能降低,心血管病风险增加。因此,青春发育期补充GH的目的是促进线性生长,降低内脏脂肪含量,增加瘦体重和BMD。但GH治疗研究的结果并不一致,尤其是男性患者(表15),可能主要与药物剂量和疗程有关。故多数建议持续给予GH治疗,直至完成青春期发育。
表15 GHD患者的青春期变化
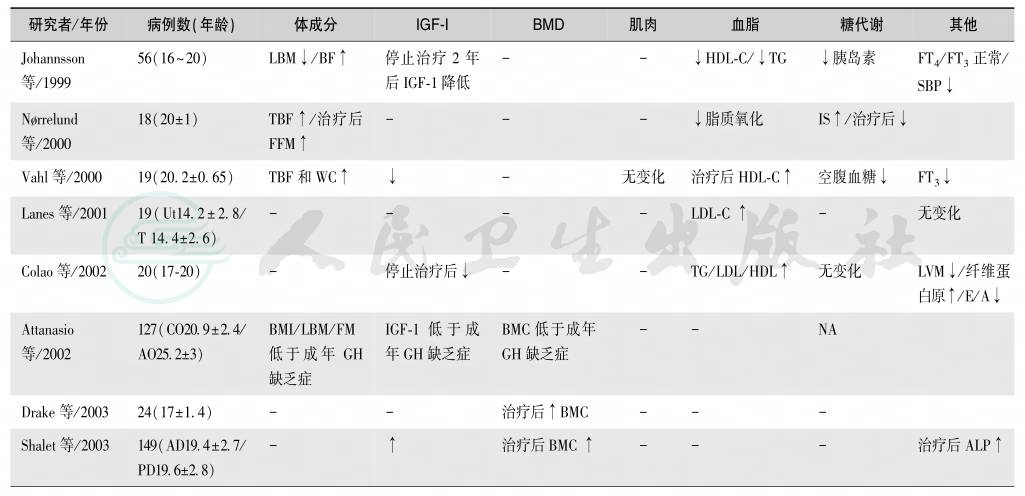
续表

注:FFM:无脂肪体质;IS:胰岛素抵抗;WC:腰围;LVM:左室质量;E/A:早期和晚期二尖瓣流速比值;BMI:体质指数;BMC:骨矿物质含量;BMD:骨密度;Ut:非治疗组;T:治疗组
2/3以上的GH缺乏患者的在经过治疗后,GH的分泌功能可转为正常(表16),但由于相关基因突变、垂体手术后或放疗后引起者难以恢复。因此,GHD在生长发育后期应该重新评价GH的分泌功能(男性14.5岁,女性16.5岁),GH分泌功能评价前需要进行GH制剂洗脱(1~3个月),一避免假阳性结果。
表16 GH缺乏患者恢复GH分泌功能情况

首先测定基础IGF-I水平,低于-2SDS者提示永久性GH缺乏症(如遗传性病变、下丘脑-垂体结果异常、获得性下丘脑-垂体病变、放疗等),高于-2SDS者应进行GH兴奋试验。一般首选胰岛素低血糖兴奋试验(切割值为GH峰值3ng/ml),或GH-精氨酸兴奋试验(切割值为 GH峰值 5~6ng/ml)。IGF-I和IGFBP-3水平有助于判断GH分泌状态。
(三)其他药物治疗
1.小剂量性腺类固醇激素
小剂量性腺类固醇激素具有GH分泌的“点火(priming)”作用。如果排除了上述因素且rhGH疗效不佳时,女性患者可试用混合雌激素(Premarin,倍美力1mg/d或雌二醇1~2mg/d),男性患者试用睾酮25~50mg/d口服。
2.芳香化酶抑制剂
骨龄的进展决定于雌激素对骨生长板的作用。在男孩中,睾酮在芳香化酶的作用下转化为雌二醇,而芳香化酶抑制剂可延长生长板的生长期,使躯体的线性生长和青春期发育延迟。在GH缺乏所致的矮小症中,骨龄进展是GH补充/替代治疗的主要障碍,芳香化酶抑制剂可阻碍躯体长高。因而该类药物常与rhGH联合应用。第三代药物可抑制体内98%的芳香化酶活性,阻滞睾酮向雌二醇、雄烯二酮向雌酮及16羟-雄烯二酮向雌三醇的转换,不良反应轻。福美坦(formestane)25~50mg/d,达峰1小时,半衰期8.9小时,最大抑制使雌二醇下降62%±14%。可用于治疗体质性青春期发育延迟、特发性矮小症和生长激素缺乏症。此外,亦可用于 McCune-Albright综合征、睾酮中毒症(testotoxicosis)和先天性肾上腺皮质增生所致的矮小症的治疗。治疗过程中应追踪骨密度(BMD)和血脂变化。该类药物对精子生成是否有影响尚无定论。
3.促GH分泌剂
促GH分泌剂及其非肽类似物可促进内源性GH释放,但其效果较GH差。葛瑞林是一种新发现的内源性GH促分泌剂,通过与垂体GHRP受体结合产生促进生长、改善代谢紊乱的作用。
4.IGF-1
美国FDA批准IGF-1用于儿童原发性IGF-I缺乏症的治疗。但是,IGF-1的最佳适应证应该是GH抵抗综合征。治疗的具体指征是身高标准差计分(height SD score)≤-3和血清IGF-1标准差计分(IGF-1SD score)≤-3的患者。 2005 年以来,已有重组人 IGF-1(rhIGF-1)供应,主要用于GH不敏感综合征和严重矮小症的治疗,但其疗效并不理想,这说明GH本身对骨骼生长板有不可补充/替代作用。rhIGF-1的主要不良反应是软组织的淋巴样增生、毛发增粗和肥胖。
5.神经递质
目前试用于临床治疗GHD身材矮小的神经递质有多巴胺和可乐定。Huseman等报道联合应用GH及L-多巴的12例GHD中,有6例生长速度比单用任一药物高。可乐定是中枢性α2受体激动剂,其主要作用可能是释放GHRH。Pintor等用可乐定治疗22例体质性生长迟缓的儿童,14例生长加速在头6个月治疗作用最大,随着服药时间的延长,生长速度减慢;有些患者停药后仍有生长加速;另一些患者停药后再次用药的生长速度减慢。体质性生长迟缓儿童常有暂时性垂体功能减退,在青春前期作GH兴奋试验时反应迟钝,在青春期发育开始后GH反应正常。
6.GnRH激动剂
GnRH激动剂(GnRH-agonist)可延长青春发育期,有利于GH的补充/替代治疗,因此有人试用GnRH激动剂加GH联合治疗,但疗效不一。
7.GHRH
GHRH治疗仅用于GH分泌障碍较轻的下丘脑性GHD患儿(严重的GHD儿童仍用rhGH治疗)。北京协和医院给15例特发性GH缺乏性矮小症患儿皮下注射GHRH1-44共 24μg/kg,共半年,14 例的生长速度由(3.1±0.3)cm/年增至(8.4±0.5)cm/年。 在 24小时内,每 3小时脉冲给予GHRH1-40,使其生长速度增快,24小时或夜间每3小时GHRH脉冲能导致下丘脑性GHD患儿的GH脉冲式分泌及生长速度增加,GHRH用量为每天 16~25μg/kg,分 2次注射。近年研制了可口服或鼻内吸入的GHRH制剂,它们的促GH分泌作用是特异的,不激活垂体的腺苷环化酶,不抑制GH的分泌。但GHRH因个别患者出现严重不良反应而于2008年停用。
(四)成年期GH治疗
成年期GHD应用GH治疗对增加瘦体重(肌肉容量)和骨量,减轻肥胖和高胆固醇血症等有一定帮助,尤其在进入成年的早期,纠正GH缺乏有重要意义。但如指征不强,要尽量避免使用。
(五)GH治疗其他生长发育障碍综合征
1.GHD伴垂体激素缺乏症
垂体激素缺乏症应同时补充相应的靶激素,如T3/T4、糖皮质激素和性腺激素等,否则GH的疗效不佳。部分GHD患者可有多发性垂体激素缺乏,因此在GH治疗的同时要监测T4、皮质醇、性激素、AVP。GH治疗可使潜在的下丘脑性甲减病情加重。若患儿对GH反应不理想,或血清T4水平降至正常值以下,应及时补充甲状腺素。确有肾上腺皮质功能减退者应长期补充可的松。必要时可给小剂量的促性腺激素或性激素以诱发青春发育。体质性生长发育迟缓者可用氧雄龙(oxandrolone),1.25mg/d或2.5mg/d,有诱导青春期发育作用,加速生长。
2.原发性IGF-1缺乏症
IGF-1治疗对GH激素不敏感综合征的患者有效。理论上推测,IGF-1也可用于 IGF-1缺乏症的治疗。目前IGF-1仅限于ⅠA型单纯性GH缺乏伴GH激素抗体及对GH治疗不敏感者。IGF-1有发生低血糖可能,使用时应严密监测血糖变化。
3.慢性肾衰竭
最佳治疗方案是肾移植。治疗目的是使血钙、磷水平恢复正常,抑制继发性甲旁亢,逆转骨骼的组织学异常,阻止和逆转软组织的钙磷沉着。在治疗原发病的基础上,还包括控制血磷、补充钙剂和维生素D。当甲旁亢症状严重时,可用骨化三醇(calcitriol)口服给药,抑制PTH的过度分泌。如PTH仍然明显升高,99mTc的摄取率无抑制,应行甲状旁腺切除术。在以上治疗的基础上,GH对慢性肾衰竭所致的矮小症的身材增高有一定作用。
4.Turner综合征
本症治疗的目的是使患者在青春期出现第二性征发育和人工月经周期,使其在心理上得到安慰。主要治疗措施为性激素补充/替代治疗,一般用雌激素/孕激素序贯治疗。一般从12岁左右开始用雌激素补充/替代治疗,炔雌醇5μg/d或己烯雌酚0.25mg/d,每月服21天,4~5个月后,在每个周期的12~21天加服甲羟孕酮5mg/d口服或黄体酮10~20mg/d肌注。2~3年后雌激素逐渐增加至炔雌醇10~15μg/d或己烯雌酚0.5~1.25mg/d。雌激素治疗促使性征发育和月经来潮。1997年,美国FDA正式批准GH制剂用于Turner综合征的治疗,但治疗剂量、疗程及长期疗效均未明确,大剂量GH治疗能使患者的身高基本达到正常。
5.Noonan综合征
性激素缺乏者可在青春期给予性激素补充/替代治疗。生长迟缓可用GH或IGF-1治疗。对已有青春期发育,但能确定进入青春期发育前期者,可试用小剂量性腺类固醇激素诱导青春期发育。









