


 收藏
收藏 已收藏
已收藏下丘脑-垂体-性腺轴的结构和功能复杂,这是该系统容易发生功能紊乱或疾病的重要原因。下丘脑性HH的病因包括遗传因素和环境因素两个方面,一般分为功能性、精神性、药物性和器质性4类,见表1。
表1 下丘脑性HH的分类

1.遗传性下丘脑性低促性腺激素性性腺功能减退症
GnRH的脉冲性分泌是下丘脑GnRH神经元的固有特征,并与多种因素有关,主要包括细胞的自发性膜电位活动、Ca2+、cAMP、GnRH受体对细胞的自分泌作用和类固醇类激素受体等。GnRH神经元细胞膜上存在同源性与异源性信号的相互作用;多种调节机制和调节途径使GnRH神经元的脉冲性分泌维持着生殖功能和性腺功能。任何原因引起下丘脑功能紊乱都可以导致HH(表2),遗传性下丘脑性低促性腺激素性性腺功能减退症致病基因见图1。
表2 遗传性下丘脑性低促性腺激素性性腺功能减退症的致病基因
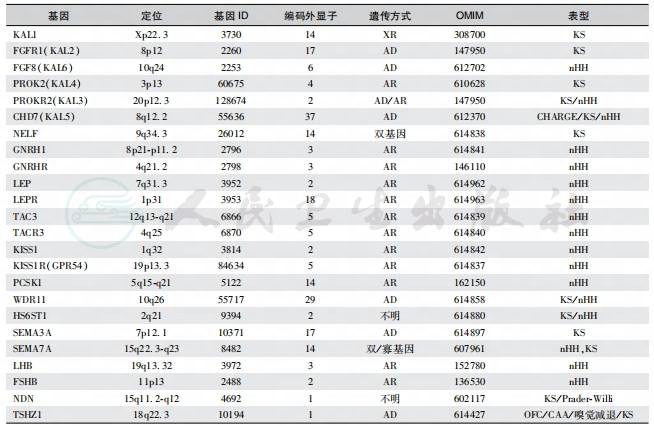
注:基因 ID:NCBI 基因数据库编号;OMIM:online catalogue of human genes and genetic disorder,人类基因与遗传性疾病在线目录号;KS:Kallmann syndrome,Kallmann 综合征;nHH:normosmic hypogonadotropic hypogonadism,嗅觉正常性性低促性腺激素性性腺功能减退症;XR:Xlinked recessive,X 性连锁隐性遗传;AR:autosomic recessive,常染色体隐性遗传;AD:autosomic dominant,常染色体显性遗传;OFC:syndromic orofacial cleft,综合征性口面裂;CAA:congenital aural atresia,先天性耳闭锁
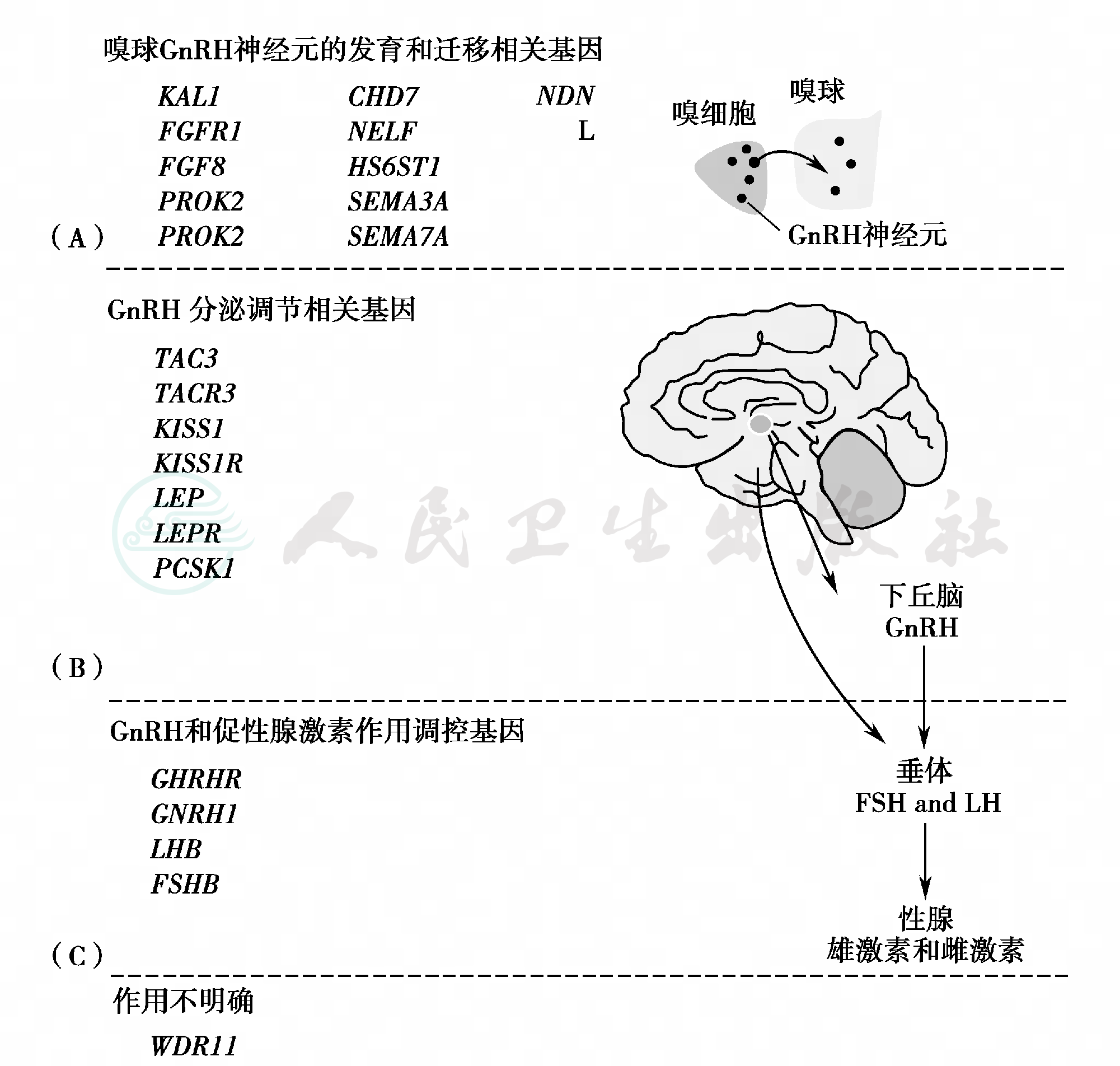
图1 遗传性下丘脑性低促性腺激素性性腺功能减退症致病基因
导致遗传性下丘脑性低促性腺激素性性腺功能减退症的致病基因作用于下丘脑垂体性腺发育和功能的不同环节。(A)GnRH神经元胚胎发育和移行血管的致病基因;(B)GnRH分泌调节血管的致病基因;(C)GnRH直接作用于垂体或间接作用于性腺的相关致病基因
下丘脑性低促性腺激素性性腺功能减退症主要包括特发性低促性腺激素性性腺功能减退症(idiopathic hypogonadotropic hypogonadism,IHH)和Kallmann综合征(Kallmann syndrome,KS)两种,其病因与G蛋白偶联受体(GPCR)的神经内分泌功能失常有关。GnRHR、KISS1R、PROKR2和NK3R突变导致GnRH缺乏、青春期发育延迟或无青春期发育;LH、FSH和性腺激素降低。在发育过程中,GnRH神经元从嗅板移行至下丘脑的正中隆突,此处的GnRH自轴突末梢释放,进入垂体门脉循环,作用于垂体,促进LH和FSH分泌。
(1)GnRHR突变:人GnRHR基因位于4q21.2,含有3个外显子,编码328个氨基酸残基的受体蛋白。GnRHR缺乏细胞内C末端尾肽,第二个细胞外环袢的191位点为赖氨酸残基具有调节受体表达和空间构象功能。缺乏该残基时,细胞表面的受体数目增加4倍,受体分子更为稳定,不容易发生内陷。目前已经发现20多种GnRHR失活性突变位点(图2),呈常染色体隐性遗传,临床表现为散发性或家族性低促性腺激素性性腺功能减退症(表3)。
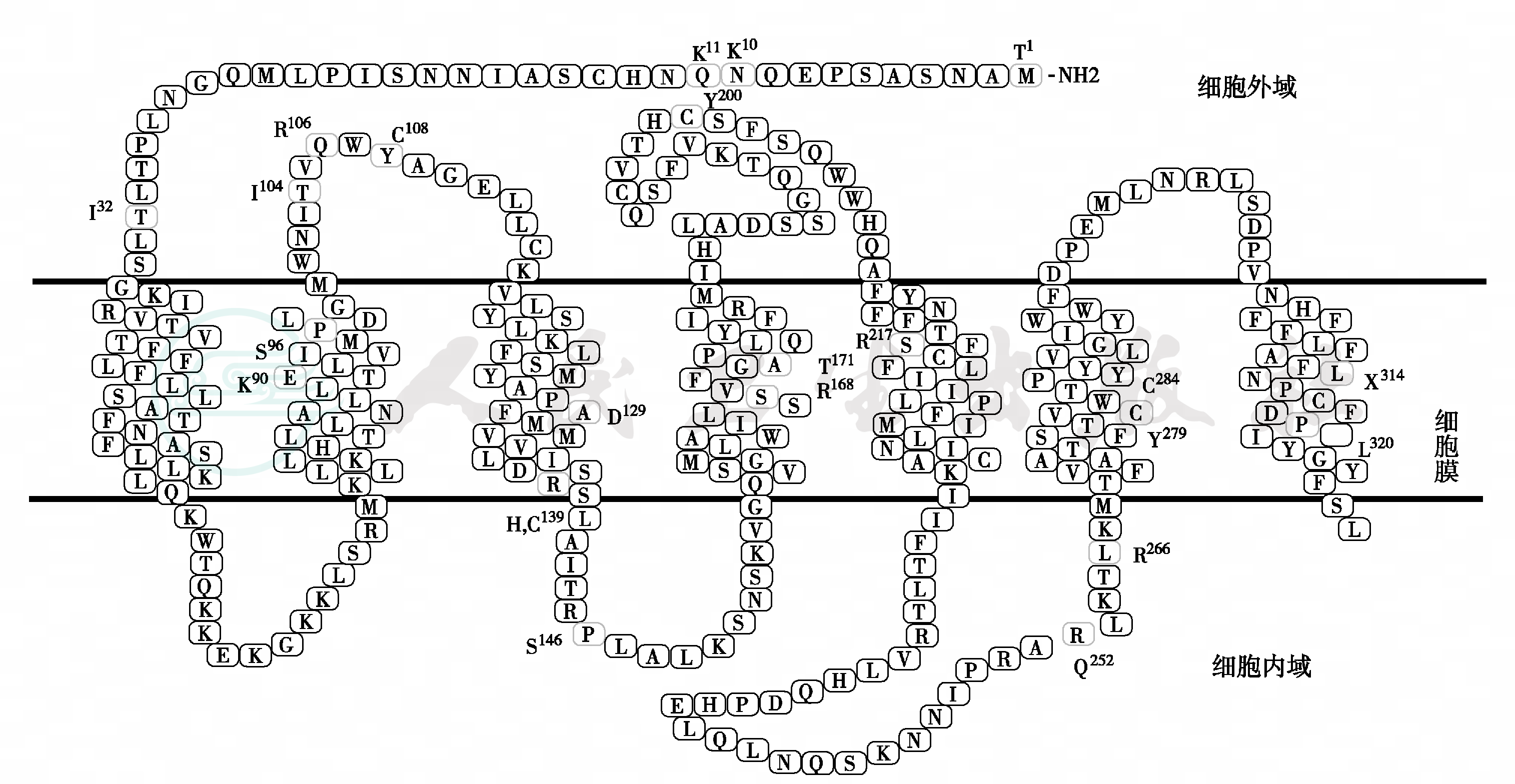
图2 GnRHR 无义突变
表3 GnRH缺乏症的基因型与临床表型
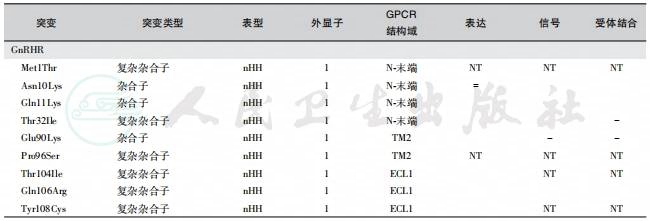
续表1
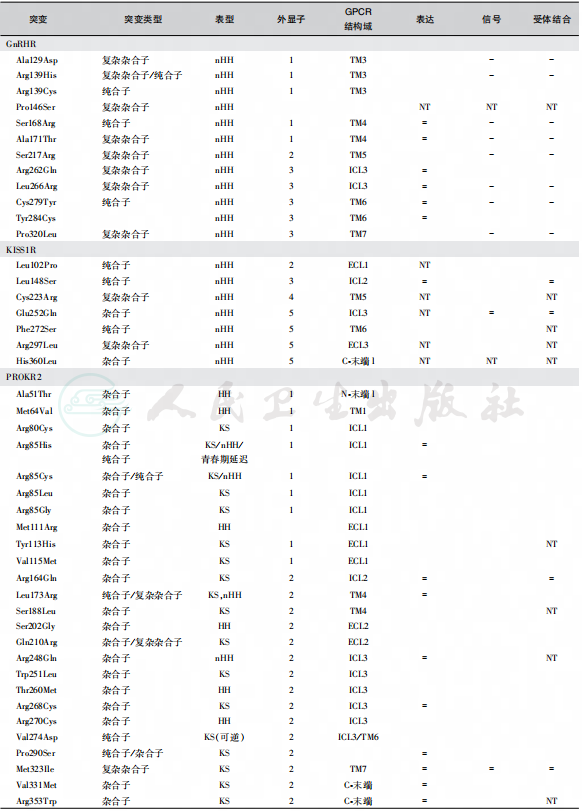
续表2
IHH:idiopathic hypogonadotropic hypogonadism,特发性低促性腺激素性性腺功能减退症;Ks:kallmann syndrome,kallmann综合征;TM transmembrane,跨膜段;ECL:extracellular loops,细胞外环;ICL:intracellular loops,细胞内环;nHH:normosmic HH,嗅觉正常性低促性腺激素性性腺功能减退症;NT:not tested,未检查;=:正常;-:缺乏
(2)KISS1R突变:目前已经有10多种突变类型报道(图3),KSS1R及其配体吻肽是调节性腺功能的重要因子,吻肽具有强烈刺激GnRH和促性腺激素释放作用。KISS1R突变引起特发性低促性腺激素性性腺功能减退症(IHH)。
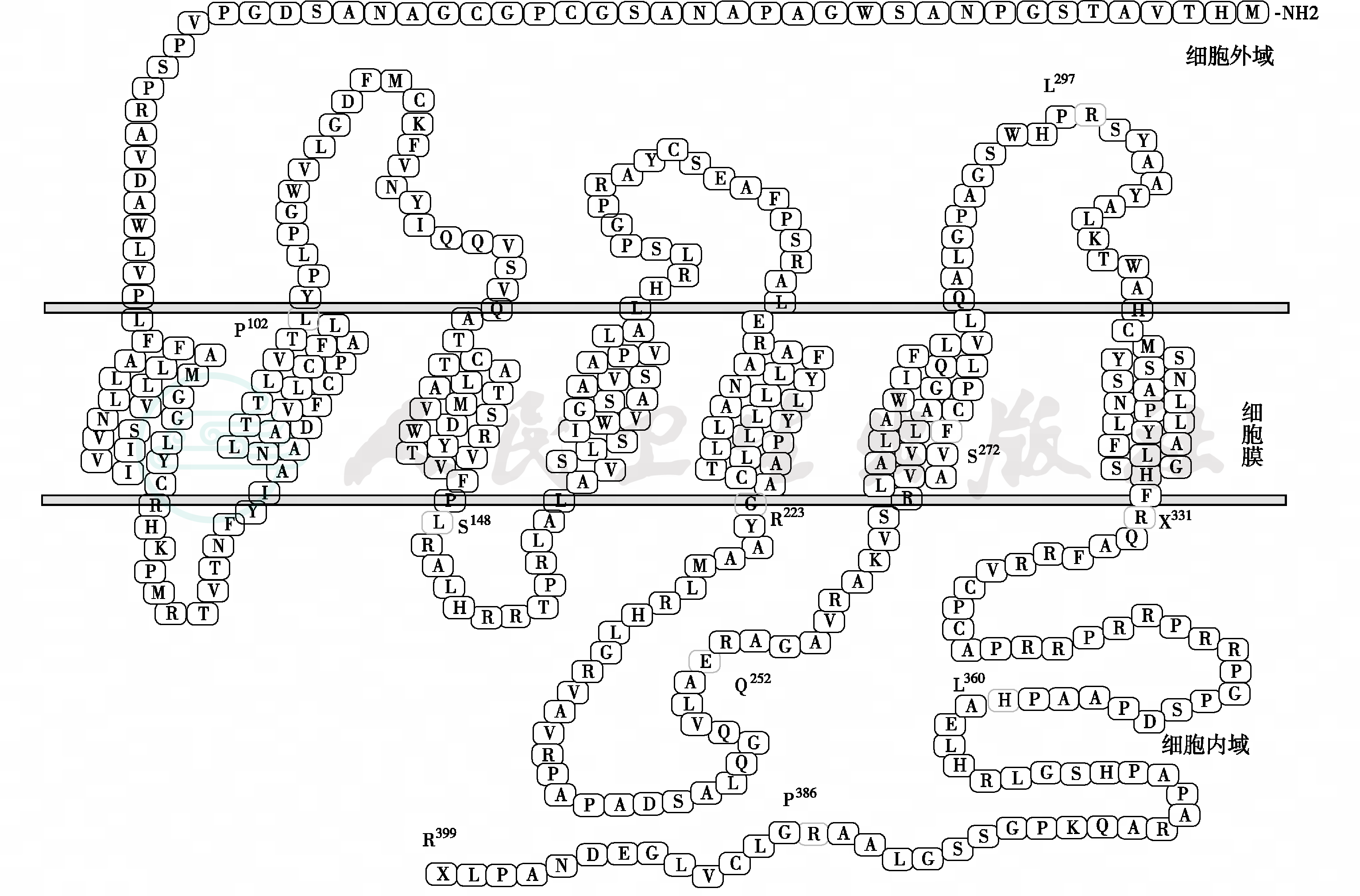
图3 KISS1R 无义突变
(3)PROKR2突变:PROK2/PROKR2系统调节肠收缩、昼夜节律、血管功能、嗅觉发育和生殖功能。2006年,首次发现PROKR2和PROK2突变引起kallmann综合征,目前报道了26种突变类型(图4)。
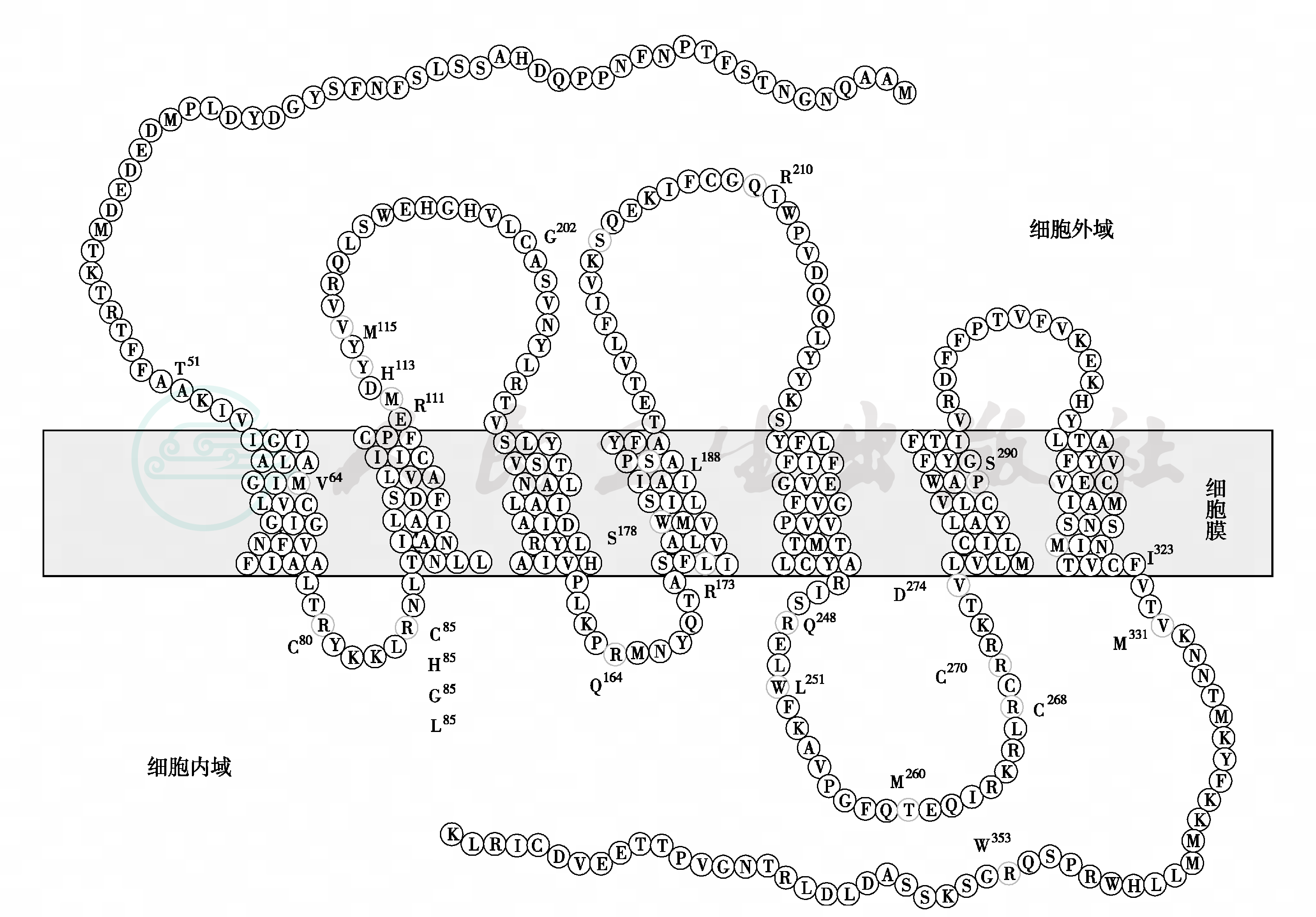
图4 PROKR2 无义突变
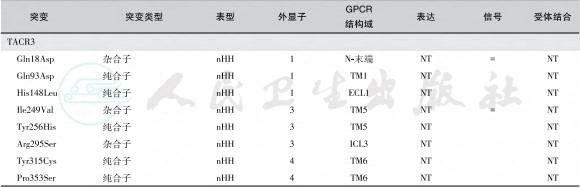
(4)TACR3突变:神经激肽B属于速激肽家族成员,速激肽家族包括P物质(substance P)、神经激肽A(neurokinin A)、神经激肽B和神经激肽K(neuropeptide K)。神经激肽B可调节脑组织兴奋性,NKB与G蛋白偶联受体NK3R结合,激活下游的IP与PKC信号通路;NK3R还能与Gs或Gi结合,引起腺苷酸环化酶的激活或抑制。TACR3亦是调节性腺功能的重要因子,因此突变后引起低促性腺激素性性腺功能减退症。下丘脑漏斗球/弓状核的神经元是形成性腺发育与功能调节的重要组分,这些神经元对性腺类固醇激素有反应,表达NKB、吻肽、强啡肽(dynorphin)、NK3R和雌激素受体ERc其功能是介导雌激素对GnRH分泌的负反馈调节,由NKB/吻肽/dynorphin/NK3R/ERc形成的调节网络具有双向、互联的特点,起着GnRH脉冲发生器(GnRH Pulse generator)作用。NKB或其受体NK3R失活性突变引起低促性腺激素性性腺功能减退症(图5和图6),患者缺乏青春期发育,血清LH和性腺类固醇激素水平降低。
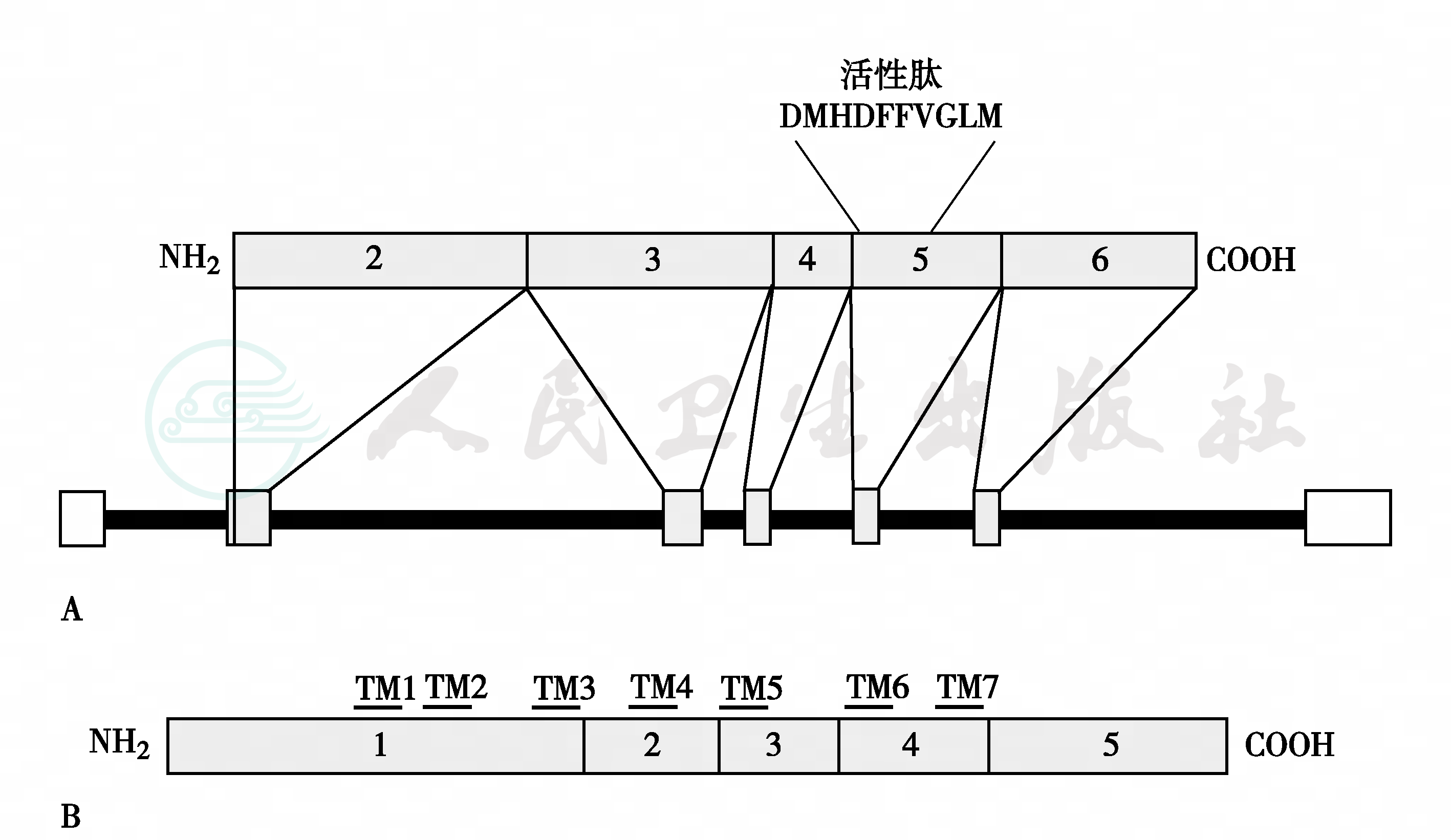
图5 TAC3 突变
A.人TAC3基因与前速激肽B原(preprotachykinin B)结构;TAC3基因含7个外显子, 外显子2~6编码前速激肽B原,外显子5编码活性NKB肽;B.NK3R G蛋白偶联受体
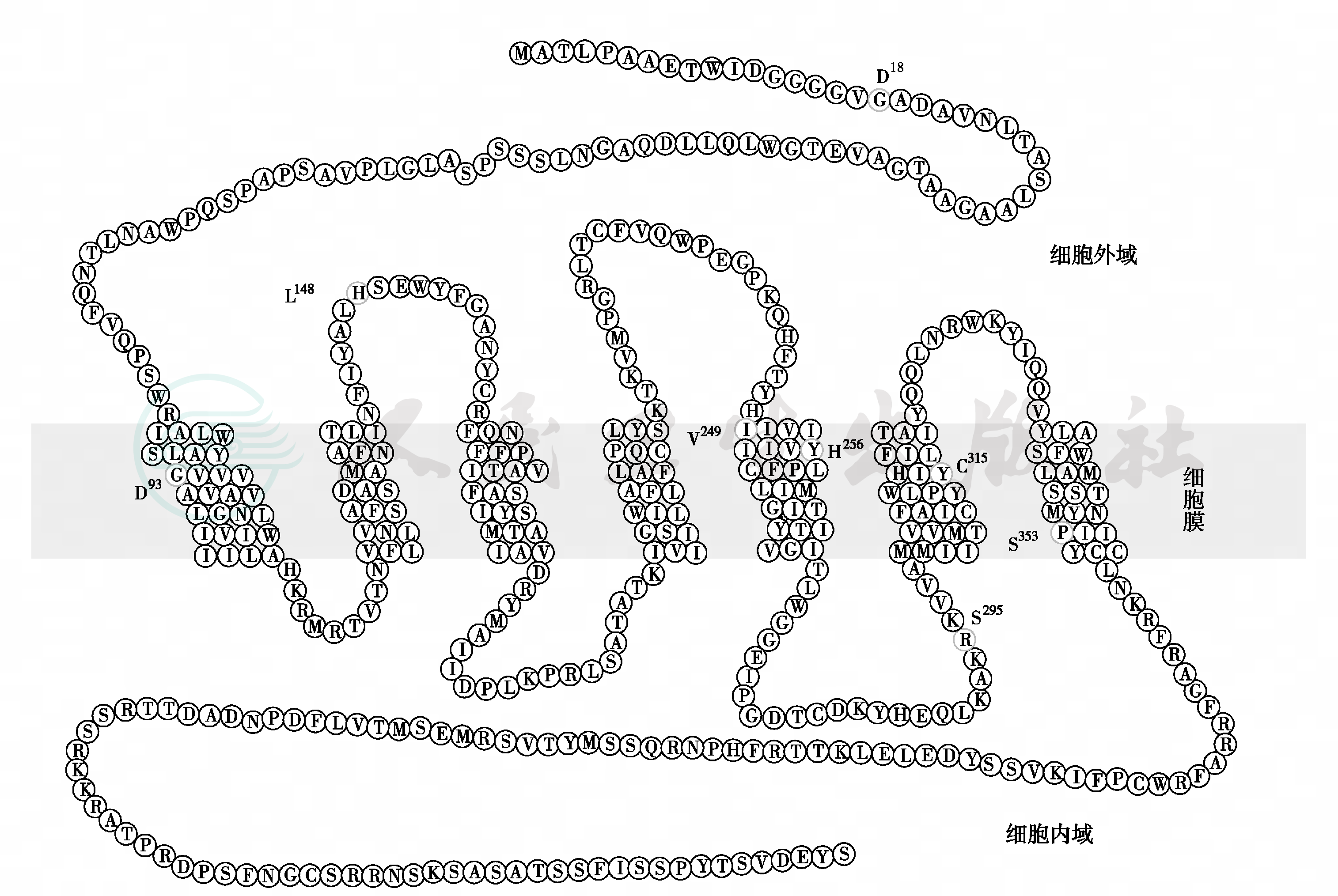
图6 NK3R 无义突变
(5)成纤维细胞生长因子及其受体突变:成纤维细胞生长因子(fibroblast growth factor,FGF)信号参与许多发育过程的调节,1型FGF受体(FGFR1)突变引起单纯性GnRH缺乏症、kallmann综合征、正常嗅觉性低促性腺激素性性腺功能减退症(normosmic idiopathic hypogonadotropic hypogonadism,nIHH),见表4。这些病例的特征是生殖功能变化不一,有的表现为严重的先天性GnRH缺乏症,另一些则仅有可逆性HH表型。
表4 FGFR1突变引起的GnRH缺乏症
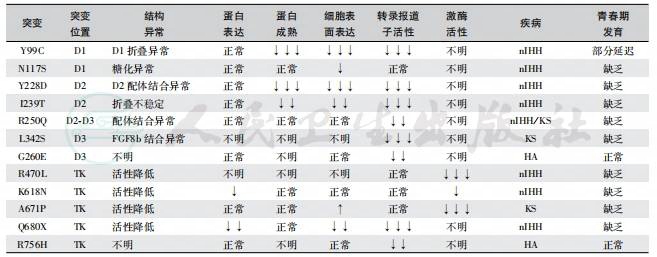
注:D1:domain 1,结构域1;D2:domain 2,结构域2;D3:domain 3,结构域3;TK:tyrosine kinase,酪氨酸激酶;nIHH:normosmic idiopathic hypogonadotropic hypogonadism,正常嗅觉性特发性低促性腺激素性性腺功能减退症;KS:Kallmann syndrome,Kallmann综合征;HA:hypothalamic amenorrhea,下丘脑性闭经;↑:mild increase,轻度升高;↓:mild decrease,轻度降低;↓↓:moderate decrease,中毒降低;↓↓↓:severe decrease,显著降低
FGFR1基因编码四种FGF受体蛋白,FGF受体为细胞表面的酪氨酸激酶受体类型,其细胞外免疫球蛋白结构域2和3(immunoglobulin domain 2/3,D2/D3)决定了其与配体的结合亲和性与特异性,C末端的D3结构域存在两种剪接方式,生成两种不同的异构体FGFR1-Ⅲb与FGFR1-Ⅲc,并由异构体调节受体反转与配体的结合特异性。上皮组织表达Ⅲb,间质组织表达Ⅲc;激活FGFR1需要分子二聚化,两个FGF分子加上硫酸肝素蛋白聚糖催化二聚体形成,使酪氨酸激酶自动磷酸化,并启动下游的信号转导(图7)。由于FGFR1突变影响的功能广泛,可出现生殖系统以外的许多临床表现(表5)。
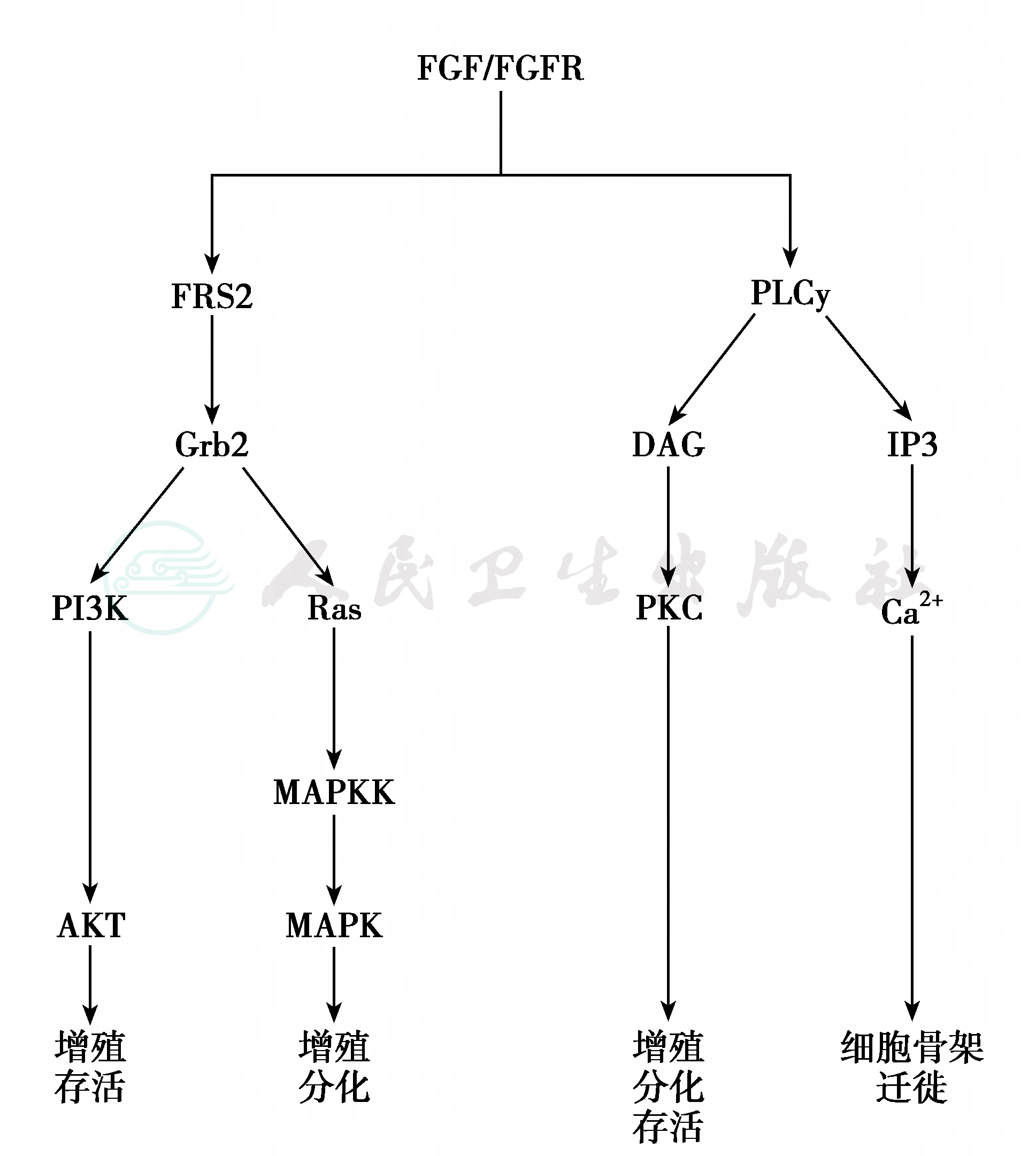
图7 FGFR 下游信号途径
FGF/FGFR结合后引起磷脂酶Cγ ( phospholipase Cγ,PLCγ)、MAPK和PI3K激活;FGF信号调节细胞 增殖与分化
表5 FGFR1突变引起GnRH缺乏症的非生殖系统表现
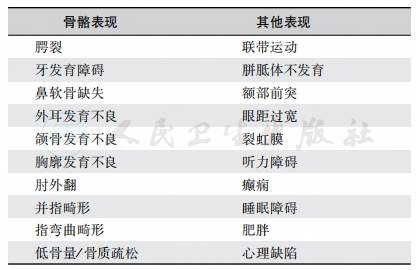
研究发现,FGF-8基因突变引起GnRH缺乏症或中线缺陷症(midline defect)。目前确定了四种FGF-8异构体,其中D94H与D73H引起脑中线缺陷症而T229M突变引起前脑无裂畸形(holoprosencephaly),见图8。
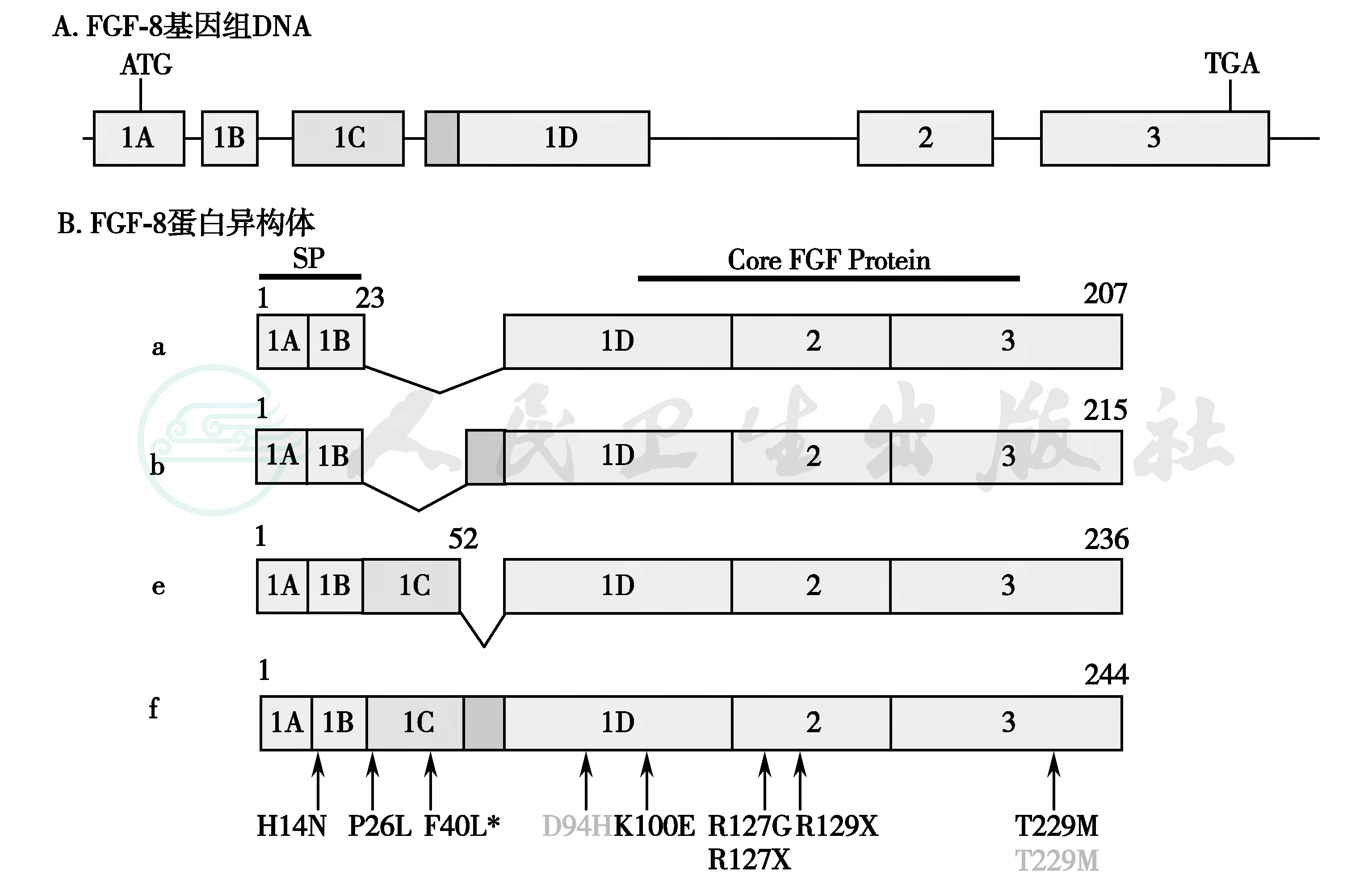
图8 FGF-8基因结构与突变位点
FGF-8基因突变引起GnRH缺乏症或中线缺陷症。A.FGF-8基因结构,方框表示外显子,线条表示内含子;B.目前确定的四种FGF-8异构体,外显子1编码1C和部分1D;FGF的保守核心由外显子2和3编码;下部为目前发现的突变位点,其中D94H与D73H引起脑中线缺陷症(如非综合征性唇裂/腭裂),T229M引起前脑无裂畸形(holoprosencephaly)
(6)DAX-突变:DAX-1基因长5kb,含2个外显子及1个内含子,编码470个氨基酸残基的蛋白质,即DAX-1。DAX-1在肾上腺及下丘脑-垂体-性腺轴表达,对其发育及功能起重要作用,在精子生成方面起关键作用。DAX-1基因的表达产物NROB1蛋白与下丘脑-垂体-性腺/肾上腺轴的发育有密切关系,突变型蛋白作为类固醇激素合成的负性调节因子而导致先天性肾上腺发育不良和低促性腺激素性性腺功能减退(图9)。AHC患者发生假性性早熟的原因与继发性ACTH过度刺激Leydig细胞ACTH受体和睾酮雄激素生成过多有关,目前发现了60余个家系的50余种DAX-1基因突变;多数是由于移码突变或无义突变导致NROB1蛋白短截。
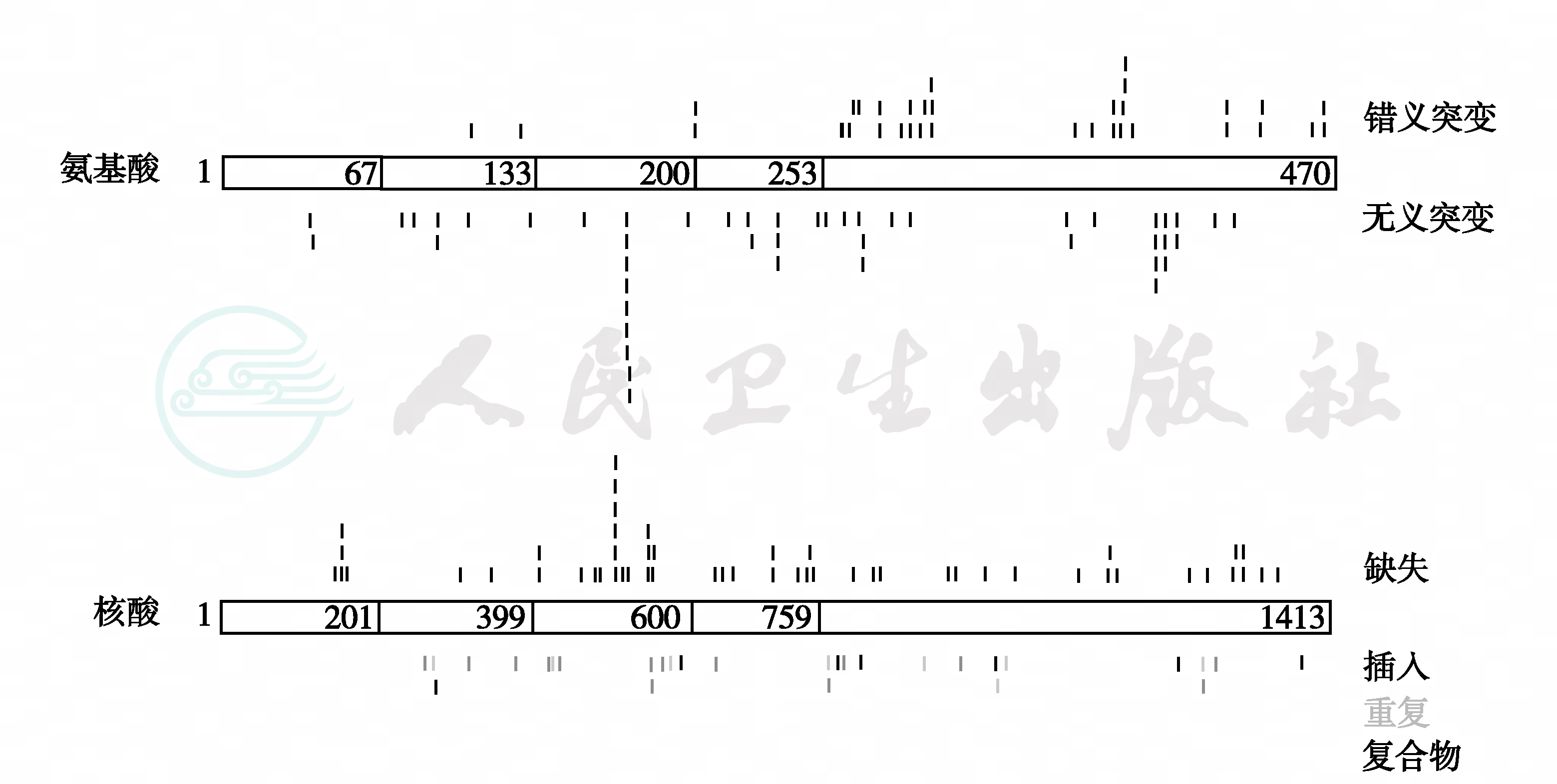
图9 DAX-1 突变
DAX-1蛋白分为三个半氨基端重复序列,即1~67、68~133、134~200和201~253四个部分及核受体样结构域254~470。上图为错义突变,下图为无义突变
2.GnIH分泌紊乱
促性腺激素抑制激素(GnIH)是新发现的下丘脑神经肽,具有抑制下丘脑GnRH、垂体LH和FSH分泌的作用。GnIH的C末端基序形式为LPXRF胺(LPX RFamide,X=L或Q),与调节痛觉的神经肽FF(neu-ropeptideFF,NPFF)的PQRF胺基序结构相似,两者来源于同一远祖基因(图10),属于含RF胺的肽类激素(RFamidepeptide)。远祖的甲状腺刺激素(thyrostimulin)含有α和β两个亚基,进化为脊椎动物后分别演变成促性腺激素(GTH)的α和β亚基,并形成异四聚体分子结构;进一步进化后,GTH的二聚体演变成腺垂体糖蛋白激素的三种功能单位,分别以不同的方式形成LH、FSH和TSH分子。
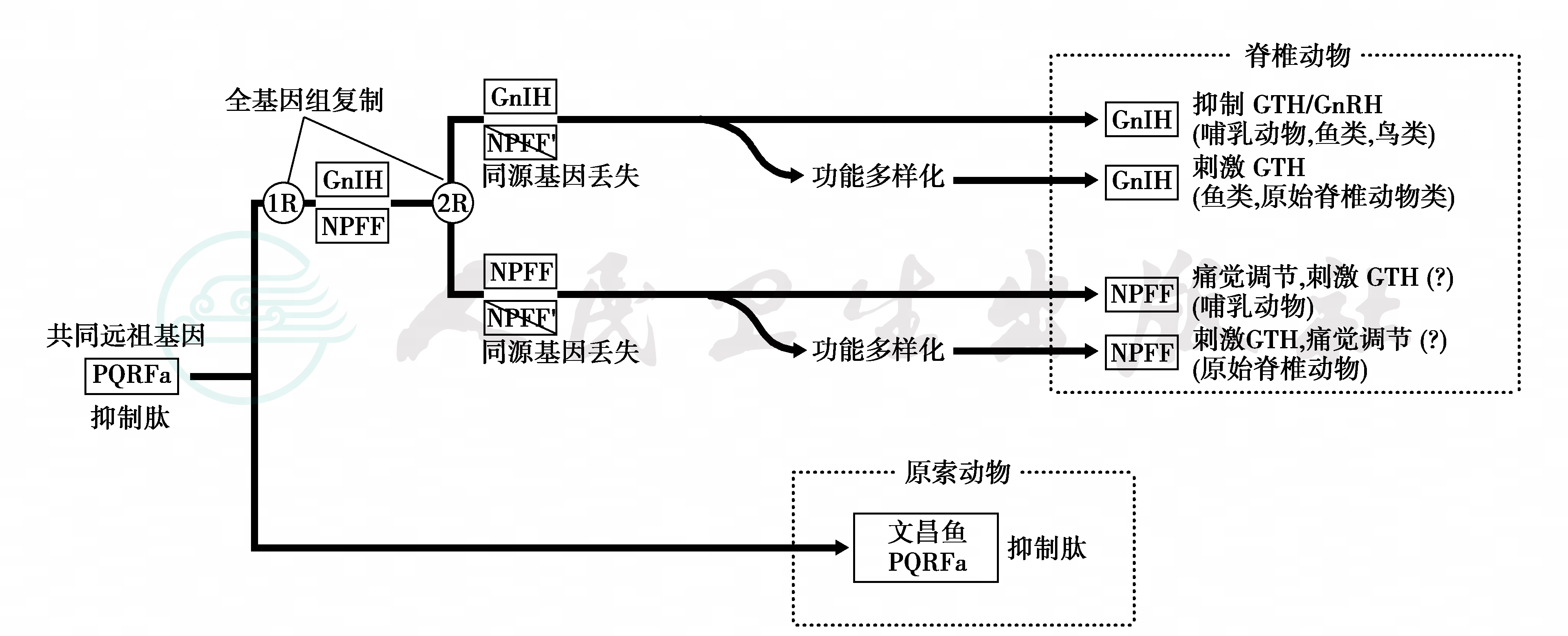
图10 GnIH与NPFF基因
GnIH和NPFF来源于共同的远祖基因PQRF;GTH包括LH和FSH
青春期发育前儿童期的GnIH生理作用是抑制下丘脑Gn-RH、垂体LH和FSH的分泌,维持性腺功能静息状态。外界环境因素(如社会应激和季节-昼夜变化)通过皮质酮或褪黑素作用于GnIH神经元,后者抑制GnRH-Ⅰ和GnRH-Ⅱ神经元活动,调节垂体LH与FSH的分泌(图11和图12),因此GnIH负性调节性腺功能。此外,GnIH也影响摄食行为。GnIH神经元与吻肽神经元密切联系,但具体功能未明。
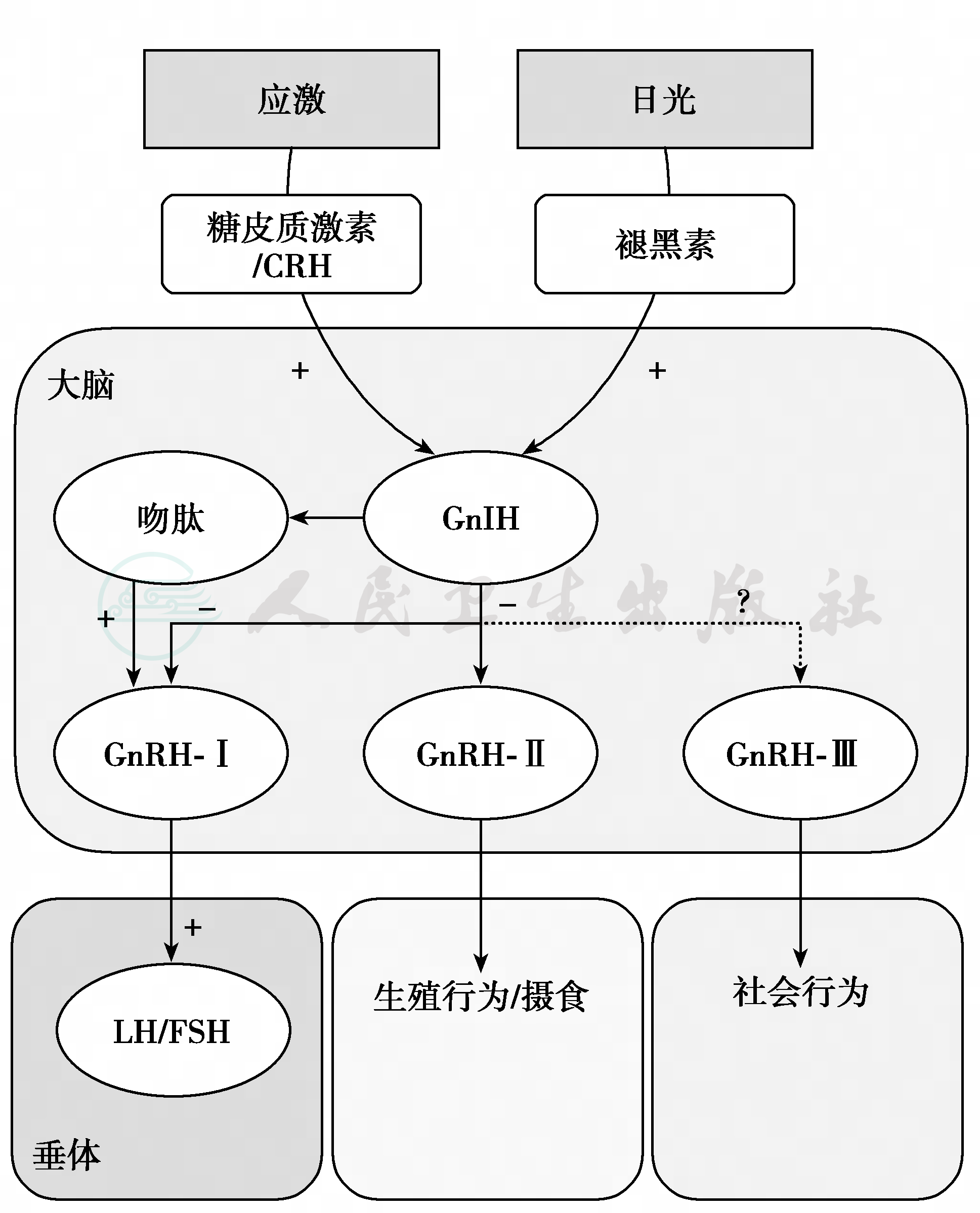
图11 环境对GnIH的作用与GnIH功能
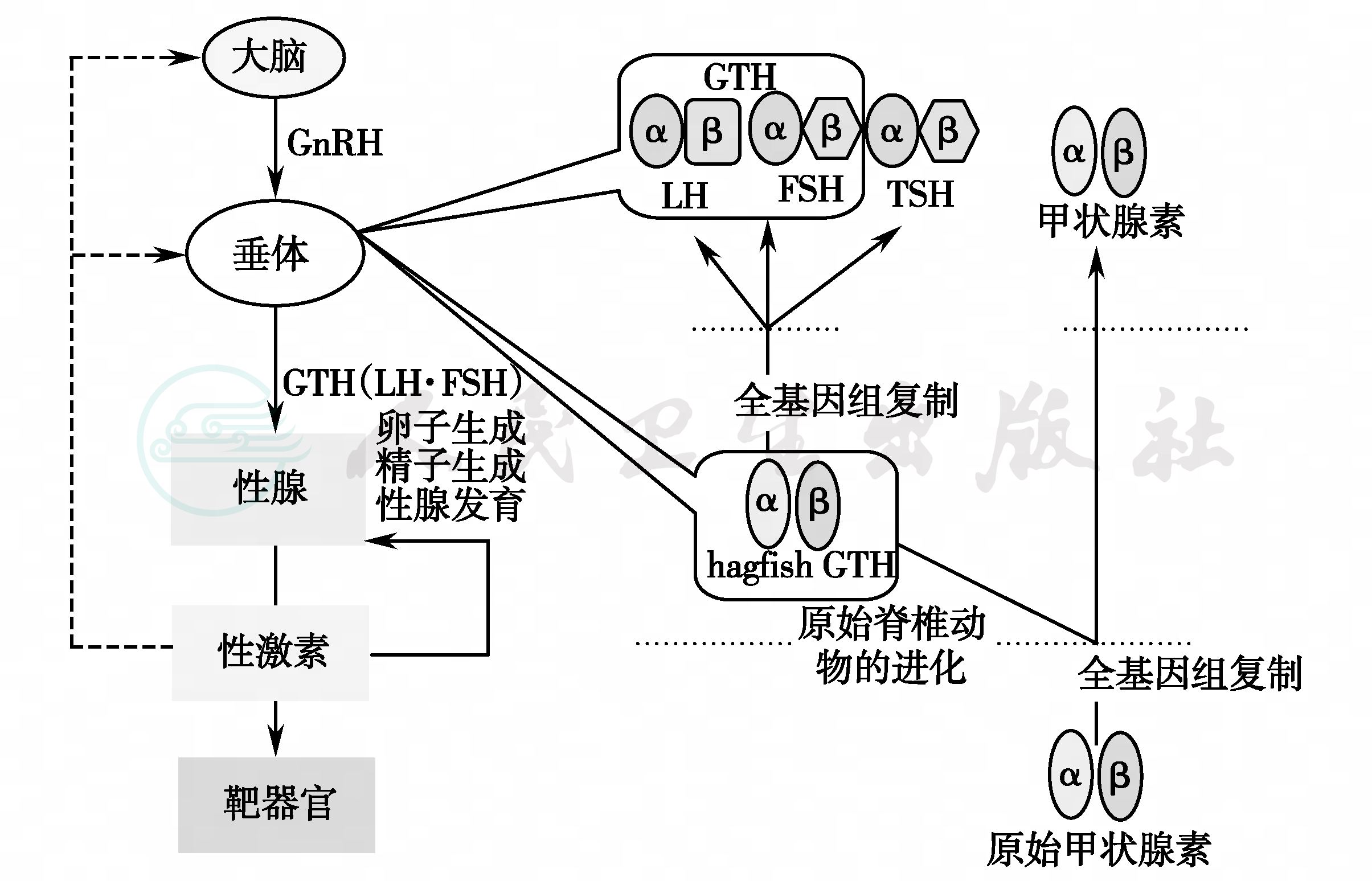
图12 下丘脑-垂体糖蛋白激素的生物进化
远祖甲状腺刺激激素(thyrostimulin )含α和β两个亚基,进化为脊椎动物 后演变成促性腺激素(GTH)的α和β亚基,在进一步进化过程中,GTH二聚体演变成腺垂体糖蛋白激素的三种功能单位,分别以不同的组合方式形成LH、FSH和TSH
3.单基因突变引起的下丘脑性HH
自从20年前首先鉴定KAL1基因突变以来,现已发现10多种引起下丘脑性HH的相关基因,部分患者甚至是双基因(digenic inherit-ance)变异所致(表6~表8)。
表6 遗传因素所致的低促性腺激素性性腺功能减退症
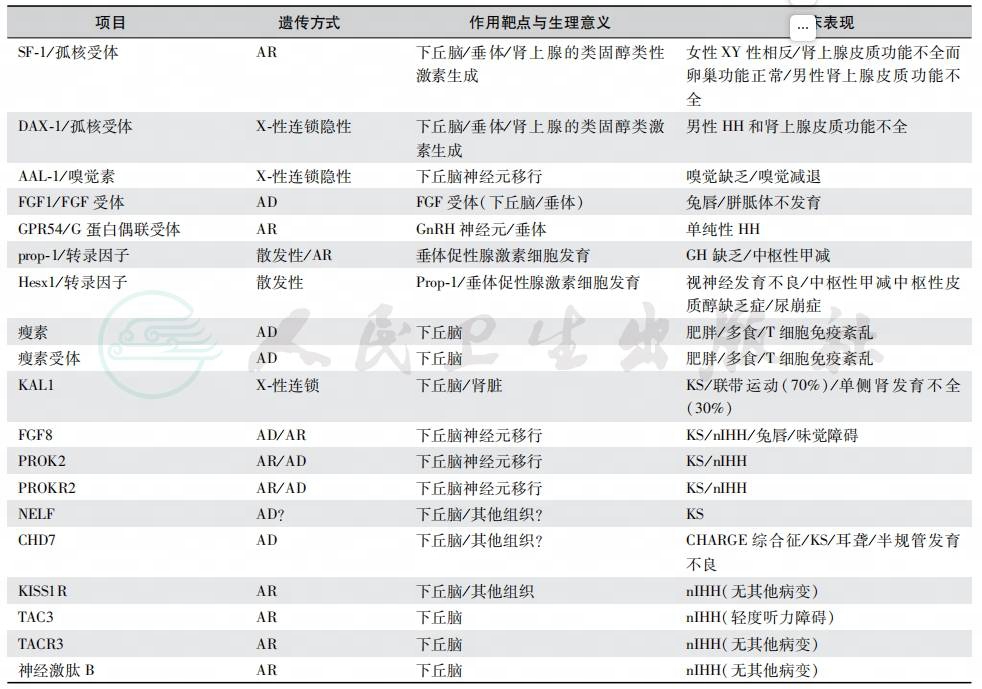
KS:Kallmann综合征;nIHH:正常嗅觉孤立性低促性腺激素性性腺功能减退症;AD:autosomal dominant,常染色体显性遗传;AR:autoso-mal recessive,常染色体隐性遗传;CHARGE(coloboma,heart anomalies,choanal atresia,retardation,genital and ear anomalies)syndrome:CHARGE综合征;NELF:nasal embryonic LHRH factor,鼻胚LHRH因子;neurokinin B:神经激肽B;嗅觉素:anosmin
表7 低促性腺激素性性腺功能减退症的双基因效应
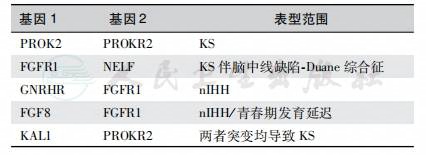
表8 低促性腺激素性性腺功能减退症的遗传性缺陷
HH:hypogonadotropic hypogonadism,低促性腺激素性性腺功 能减退症;CHARGE:colobomaa-heart anomalies,choanal atresia,retarda- tion-genital and ear anomalies,缺失-心脏畸形-鼻后孔闭锁-发育延迟-生殖器与耳畸形;KISS1R:吻肽1 receptor,吻肽1受体
4.获得性下丘脑性低促性腺激素性性腺功能减退症
(1)特发性低促性腺素性性腺功能减退症:以前的特发性HH(IHH)专指由于GnRH缺乏引起的HH。如上所述,其病因和发病机制已经基本明了,主要因相关基因(如KAL1、GnRHR、FGFR1、GPR54、PROK2、PROKR2、FGF8、CHD、TAC3、TAC3R等)突变所致。但在临床上,因为患者缺乏嗅觉障碍而被臆断为IHH。此处所述的IHH不包括已经明确病因的遗传性单基因突变者。由于患者下丘脑分泌GnRH的神经元缺如或数目减少与功能降低,或因Gn-RH功能异常导致血LH、FSH、睾酮(或雌二醇)降低,伴青春期发育延迟及性腺和第二性征不发育。女性无月经,乳腺细小,阴毛少;男性睾丸和阴茎均小,阴毛、腋毛稀少,喉结不明显,但不伴嗅觉障碍,也无器质性颅内病变或垂体疾病。
血LH、FSH及睾酮降低或测不出。注射GnRH后,血LH及FSH仍低或无反应;但经数日多次注射后,出现LH/FSH升高反应甚至超高反应。用此试验可鉴别垂体疾病所致的低促性腺激素性性腺功能减退症。氯米芬可在受体部位与雌二醇竞争性对抗,口服氯米芬(每次50mg,每天1~2次,连续14~30天)后无反应,而正常人的血LH明显升高。外源性GnRH替代是本综合征的最佳治疗方案。在无GnRH供应或患者FSH缺乏不严重时,可用睾酮类制剂或单用HCG治疗,以促进和维持性腺功能及第二性征。对较重的患者或为了促进生育力,应每周加用HCG 2000~4000U,分3次肌内注射,长期治疗可获得性成熟。如未达到目的,可改用人尿促性腺激素(menotrophin,HMG),每周肌内注射3次。
生育后再改用睾酮维持性腺功能,无效时再改用重组的人FSH或LH治疗。
(2)继发性低促性腺素性性腺功能减退症:部分HH(如Fröhlich综合征)是由于脑部器质性病变所致,如脑肿瘤、脑炎、小头畸形、脱髓鞘等,但腺垂体的其他功能正常。还有一些综合征伴有下丘脑性肥胖及性腺功能减退(多为遗传性)和中枢神经功能异常,如Laurence-Moon综合征、Bar-det-Biedl综合征、Allstrom-Hallgren综合征、18-三体综合征(Edwards syndrome)和Prader-Willi综合征等。获得性中枢神经系统疾病引起下丘脑-垂体功能障碍,如Hand-Schüller-Christian病累及下丘脑时,常出现尿崩症和其他下丘脑功能紊乱的临床表现。Prader-Willi综合征和Angelman综合征是由于丢失15q11-q13所致。Prader-Willi综合征患者常伴有肌张力低下、进食困难,以后发生多食、肥胖、短肢和HH,智力仅轻度异常;Angelman综合征伴有严重智力障碍和畸形。全身性疾病与药物引起的HH见于结核、结节病、其他感染性炎性病变、血管病变及创伤等累及下丘脑或垂体时。大约1/3的男性2型糖尿病患者伴有低促性腺激素性低睾酮血症;同样的情况亦见于代谢综合征及肥胖,但较少见于1型糖尿病。伴有情绪障碍(特别是双相情感障碍)的女性容易并发生殖功能紊乱,部分伴有HH,其原因未明,可能与长期应用抗精神病药物有关。此外,阿片中毒和长期耐力锻炼亦可引起HH。
一些肾上腺皮质增生由于ACTH过度分泌,肾上腺皮质分泌大量雄激素,或者长期使用大剂量外源性雄激素/雌激素,可抑制下丘脑-垂体的GnRH-LH/FSH系统,引起低促性腺激素性性腺功能减退症。如果持续的时间够长,可进一步导致继发性睾丸不发育与纤维化。DHEA、DHEAS、雄烯二酮以及它们在外周组织转换的睾酮能维持男性化,但亦促进中心性肥胖与脂肪肝的发生,增加心血管疾病的风险。这些患者虽然男性化正常,而睾丸细小,坚硬,受孕能力降低或缺乏生育功能,但对GnRH-LH/FSH刺激有反应,并可用于该种HH的治疗。女性高雌激素血症也存在类似情况。青春期发育延迟可继发于许多慢性疾病及长期应用糖皮质激素者。青春期发育延迟可导致低骨量,多伴有牙发育延迟。
常见的下丘脑性HH有三种临床类型。①Kallmann综合征:下丘脑性HH伴嗅觉减退或嗅觉缺失(hyposmia/anosmia);②肥胖-生殖无能综合征(syndrome of adiposogenital dystrophy):下丘脑性HH伴明显肥胖,系某些病变累及下丘脑,导致神经-内分泌功能紊乱,从而引起GnRH分泌减少和继发性性腺发育不良与脑性肥胖;③下丘脑性无排卵(hypothalamicanovulation):是指下丘脑性HH引起的无排卵与不育症,继发性垂体LH/FSH缺乏引起的HH病因不在垂体而在下丘脑。
1.低促性腺激素性性腺功能减退症
目前已经确定的HH病因有:①GnRH受体1突变;②LHβ链突变;③DAX1突变(伴有肾上腺发育不良);④瘦素/瘦素受体突变(伴有肥胖);⑤激素原转化酶1(prohormone convertase 1)突变;⑥吻 肽受体(吻肽receptor,GPR54)突变;⑦KAL1(编码anosmin-1)突变(X-性连锁性Kallmann综合征);⑧KAL2(编码FGFR1)突变(常染色体显性遗传性Kallmann综合征);⑨他垂体发育因子如鼻胚LHRH因子(nasal embryonic LHRH factor,NELF;基因Nelf)、PKR2、CHD7等基因突变。
2.CHARGE综合征
NELF参与Gn-RH细胞和嗅觉神经元的移行,但目前仍不明确NELF的具体功能。应用外源性GnRH后,垂体的NelfmRNA表达升高3倍以上,因而NELF可能是一种垂体发育转录因子。proki-neticin2(PROK2)或其受体(PROKR2)是表达昼夜节律信号的中介因子,与生物钟的关系密切。眼缺失-心脏畸形-后鼻孔闭锁-生长发育迟缓-泌尿道畸形-耳畸形-聋哑综合征(CHARGE综合征)是编码染色体结构域螺旋酶DNA结合蛋白7(CHD7)突变所致的遗传性疾病。
3.Kallmann综合征
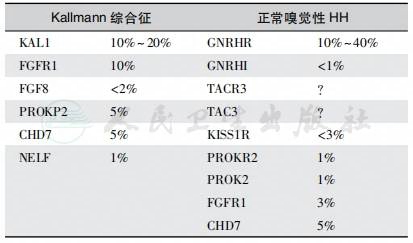
Kallmann综合征为最常见的低促性腺素性性腺功能减退症,伴嗅觉失敏(或丧失)和神经缺陷(神经性耳聋及色盲等)。主要病变在下丘脑及邻近的嗅觉中枢,GnRH分泌不足是本病的发病原因。若下丘脑的功能异常广泛,可伴发渗透压受体功能异常及渴感异常。该综合征可呈X连锁隐性遗传、常染色体显性遗传或常染色体隐性遗传。大约60%的HH患者伴有嗅觉缺失,其程度不一,极轻者往往被忽视而臆断为IHH。主要与KAL1或成纤维生长因子受体1(fibroblast growth factor recep- tor1,FGFR1)基因突变有关,但只占所有Kallmann综合征病例的15%,另外15%的病例是由于PROK2或PROKR2突变所致。在40%嗅觉正常的IHH患者中,部分存在GnRH、KISS1R、TAC3、TACR3突变。有些基因突变(如PROK2、FG-FR1或SOX2)既可引起Kallmann综合征,亦能导致IHH。
4.特发性低促性腺激素性性腺功能减退症
吻肽和神经激肽B(neurokininB)是激发GnRH分泌并促进性腺成熟的调节因子。神经激肽B受体由TAC3基因编码。主要分布于下丘脑,调节GnRH分泌,因而神经激肽B或其受体突变是下丘脑性HH的重要原因。吻肽及其受体(GPR54,KISSR)是调节生殖功能的关键因子,突变后引起特发性HH。IL2~10可引起kiss1r结构重排,提示一些分子与吻肽的GPCR/G蛋白信号存在“对话”现象,而KISS1R的活化性突变(Arg386Pro)又可以导致性早熟。吻肽受体突变所致HH的特点是:①突变位点遍布受体的全段,没有“突变热点”;②一般不伴嗅觉缺失(Kallmann综合征)、颅面中线缺陷和骨骼异常;③缺乏青春期发育而生殖系统正常;④LH分泌脉冲存在,振幅降低,但患者对内源性或外源性GnnRH有反应;⑤KISS1R的活化性突变(如R385P)引起中枢性性早熟。
5.纤毛病相关性遗传综合征
细胞器中心粒(centriole)为桶状结构,是形成纤毛(cilia)与鞭毛(flagella)的必需元件。中心小体(centrosome)异常导致基因组不稳定,并可诱发肿瘤。中心小体蛋白突变引起小头畸形、矮小症与HH,而中心粒重排引起纤毛病(ciliopathy)。
6.Lowe综合征
又称为X-性连锁遗传性眼-脑-肾综合征(X-linked oculocerebrorenal syndrome),其特征是发育延迟、失明、肾小管功能障碍伴进行性肾衰竭,病因为编码磷酸酶的OCRL基因突变。该基因突变引起Dent病(Dent病伴轻度Lowe综合征表型)。Joubert综合征的表现是小脑共济失调、色素性视网膜病(pigmentary retinopathy)和肾病,病因为编码纤毛蛋白的相关基因(至少已经发现5种)突变。Bardet-Biedl综合征除智力障碍、HH、视力下降和肥胖外,亦伴有肾囊性变或囊性纤维化,已经确定的12个致病基因均与纤毛蛋白功能障碍相关,如Meckel综合征。
7.下丘脑无排卵
下丘脑GnRH神经元合成GnRH并以特有的脉冲方式分泌到垂体门脉中,调节垂体FSH/LH的释放。GnRH神经元的活动受性腺类固醇激素和中枢性神经递质(如去甲肾上腺素、多巴胺、β-内啡肽等)的调节,因此,任何影响中枢神经功能的因素都可能导致GnRH分泌异常。同时,性腺类固醇激素过多或过少亦干扰GnRH的合成和分泌,部分患者无月经或月经稀少。
(1)功能性下丘脑性无排卵:GnRH分泌异常导致下丘脑性无排卵,其病理生理特点是在没有卵巢被抑制(如过量雌激素和抑制素)的情况下,GnRH缺乏正常的高峰分泌脉冲,代之以低幅的GnRH分泌频率,从而导致LH分泌异常。GnRH的分泌脉冲各不相同,绝大多数患者的LH分泌模式与青春期前类似,少数较严重患者的LH/FSH分泌脉冲和频率均明显降低,但对外源性GnRH的反应仍正常。下丘脑性无排卵多发生于脑力劳动者、长跑运动员、芭蕾舞演员或精神受到突然刺激者,以年轻未婚女性多见,可有突然精神刺激、剧烈运动、过度恐慌、忧郁等病史。
(2)器质性下丘脑性无排卵:以特发性HH伴嗅觉障碍(Kallmann综合征)最为典型,患者的青春期发育延迟为不可逆性,并可导致不育和不孕。
1.病因治疗
一般驱除原发因素(如肿瘤、炎症等)后可恢复月经和排卵。无生育要求和性激素水平特别低下的患者可给予雌激素(女性)或雄激素(男性)替代治疗。短期雄激素替代治疗可改善代谢,提高胰岛素敏感性,增加肌肉含量;长期雄激素替代治疗可改善患者的生活质量(包括婚姻状态、性生活质量)和机体代谢(包括糖尿病发生、血脂、机体脂肪含量)状态。HH患者有生育需求时,可以从睾酮替代治疗切换到生精治疗方案。
2.促生精治疗
其原理是模拟FSH和LH对睾丸的刺激作用。如果女性患者要求恢复排卵和生育功能,应给予GnRH治疗。因为重组的人FSH与LH的疗效满意,所以GnRH治疗的地位较以前有所下降。lutropin-α是重组的人LH,每日皮下注射75U即可。一般应在卵泡刺激前半个周期,FSH/LH的比例为2∶1;卵泡刺激的后半个周期的比例为1∶2。有生育要求的男性HH可用GnRH治疗,经过一段时间的治疗后,如果睾丸仍然细小,可用重组的人FSH治疗,以恢复精子数目。研究发现,男性HH患者应用FSH/ LH联合治疗的效果亦较佳。特发性HH的治疗按LH分泌的生理频率与幅度,脉冲式给予外源性GnRH,每90分钟给1次脉冲注射(含GnRH 5μg),总疗程6~12个月,每个脉冲的GnRH用量根据个体对治疗的反应酌情调整。如无GnRH,也可用HCG或HMG代替,但易引起超排卵与流产。
根据北京协和医院内分泌科的经验,垂体瘤术后患者经生精治疗后,精子生成的成功率接近100%,IHH患者的精子生成率低,而先天性的腺垂体功能减退症患者的成功率更低。
3.促性腺激素治疗
GnRH类似物是治疗HH和中枢性性早熟的革命性进步。对发病<6岁的女性性早熟患儿增高疗效确切。由于脉冲性GnRH治疗泵不甚方便,常选用促性腺激素治疗女性低促性腺激素性性腺功能减退症。每天注射促性腺激素(HMG、FSH、LH)诱导排卵的效果与GnRH相当或稍差。常用剂量75U/d,排卵率80%~100%;与LH联合治疗的妊娠成功率明显高于单独FSH者。FSH与LH、HMG治疗的主要风险是发生卵巢过度刺激综合征和多胎妊娠。









