


 收藏
收藏 已收藏
已收藏中文别名 :SSc合并肺间质纤维化
SSc肺部病变最常见的是间质性肺疾病(ILD),ILD在局限性皮肤型或者弥漫性皮肤型SSc均可发生。SSc患者肺活检有肺纤维化表现者高达70%~80%。SSc合并肺间质纤维化和IPF的病理表现很难鉴别。早期改变包括间质水肿,肺泡壁增厚,肺泡间隔内炎症细胞浸润(单核细胞、中性粒细胞、嗜酸性粒细胞和淋巴细胞等),结缔组织基质细胞浸润,成纤维细胞增生,胶原蛋白沉积,以及肺泡腔内炎症(主要是巨噬细胞),Ⅱ型肺泡细胞增生,不同程度的血管腔闭塞。病变主要分布在胸膜下、肺底及肺后段。由于广泛的纤维化导致肺泡壁逐渐变薄、破裂,形成许多小气囊。还可能有肺动脉高压表现。SSc相关性肺间质纤维化者病理改变以非特异性间质性肺炎(NSIP)类型为主,明显多于普通型间质性肺炎(UIP)的类型。SSc相关性间质性肺炎中,NSIP类型占77.5%,UIP类型占7.5%,终末期病变不能分类的有7.5%,其他类型包括呼吸性细支气管炎伴间质性肺疾病(RB-ILD)、结节病、机化性肺炎等。UIP病理学特点为病变在空间分布和时间分布上都存在明显的异质性:正常肺组织和病变组织间杂;不同部位的病变新旧程度不一,有些病灶处于活动性炎症期而另外一些则处于明显纤维化期,可以见到活跃增殖的成纤维细胞灶和胶原沉积或者蜂窝样改变同时存在。而NSIP特点为肺泡壁内有不同程度的炎症或者纤维化改变,但在时间上基本一致,无UIP新旧病灶共存的现象。
SSc合并肺间质纤维化的发病机制并不十分清楚。
(一)血管内皮在SSc肺纤维化中的作用
SSc病程早期,激活的、表达高水平Ⅰ型和Ⅲ型胶原蛋白mRNA的成纤维细胞出现在血管附近,提示血管相关事件的发生介导了成纤维细胞的活化及组织的纤维化。毛细血管内皮的损伤,导致血管通透性增加。SSc患者支气管肺泡灌洗液中白蛋白、IgG、中性粒细胞弹性蛋白酶和胶原酶浓度增高,证明了毛细血管渗漏的存在。SSc患者肺核素显像时99mTc-DTPA清除率增高,提示肺毛细血管通透性增加。肺血管通透性增加导致炎症细胞和血浆蛋白渗漏到肺间质,从而刺激成纤维细胞增生、胶原合成。另外,血管内皮细胞本身可以产生具有成纤维活性的因子,血管内皮表达或产生内皮素-1(ET-1)增加,肺泡炎症细胞释放的细胞因子(TNF-α、TGF-β、IL-8等)可能介导了这一过程。ET-1是一种可以收缩血管和促进有丝分裂的多肽,在胶原合成及纤维化过程中起重要作用。另外,SSc毛细血管内皮的血小板衍生生长因子(PDGF)表达增加。ET-1、PDGF可能与其他细胞因子以及生长因子协同激活了成纤维细胞。
(二)肺泡炎
免疫反应触发炎症过程,在SSc肺泡细胞内层液体里存在免疫复合物。在肺损伤之前就已经存在肺泡炎,大量的免疫和炎症细胞(包括活化的巨噬细胞、中性粒细胞、嗜酸性粒细胞和淋巴细胞等)在肺泡结构内聚集,这可能是肺纤维化的第一步。炎症过程导致了肺泡细胞以及细胞外基质的损伤,从而启动了修复过程。巨噬细胞、嗜酸性粒细胞和中性粒细胞可以通过释放活性氧或者蛋白溶解酶来损伤肺组织,SSc的肺泡巨噬细胞和正常的巨噬细胞比起来能释放更多的超氧阴离子,并且SSc患者BAL中的胶原酶和中性粒细胞弹性蛋白酶是增高的。淋巴细胞可以产生免疫球蛋白,继而形成免疫复合物从而导致肺损伤,并且可以通过减少产生抑制成纤维细胞增殖的细胞因子(如IFN-α、IFN-β、IFN-γ等)而促进肺纤维化。炎症细胞还可以激活凝血系统,表现为BAL中的纤溶酶原激活物水平增高。炎症细胞可以产生趋化因子或者通过调节血管内皮和白细胞黏附分子的表达来募集炎症细胞,进一步促进炎症反应。炎症细胞产生的细胞因子和趋化因子还使得成纤维细胞募集及沉积在肺内,生成结缔组织基质。SSc患者的巨噬细胞释放大量的IL-1、IL-6、TNF-α、纤维连接蛋白及肺泡巨噬细胞起源的生长因子。SSc相关ILD患者肺泡巨噬细胞的IL-8 mRNA和蛋白水平也增高。
(三)肺纤维化形成
纤维化过程是生长因子和细胞因子调节的结果。生长因子和细胞因子通过趋化作用使成纤维细胞聚集到炎症部位,并且可以调节成纤维细胞增殖和基质合成。单核因子直接刺激成纤维细胞、或者通过诱导生长因子来刺激成纤维细胞增殖,还可以通过合成前列腺素 E2 (PGE2)、IFN-α、IFN-β、IFN-γ、TNF-α等来抑制成纤维细胞生长。这些因子的失衡可导致Ⅲ型胶原合成增加,并在肺内沉积。巨噬细胞为主的炎症细胞释放大量的生长因子和细胞因子从而改变成纤维细胞的增殖和胶原的合成。SSc患者BAL中的肺泡巨噬细胞分泌大量的FN,FN是成纤维细胞的生长因子,也是间叶细胞的趋化因子。TGF-β、胰岛素样生长因子和PDGF也是肺泡巨噬细胞的产物,具有成纤维活性,在SSc患者的BAL中也是增高的。这样,细胞因子通过刺激或者抑制作用调节了SSc成纤维细胞活性,在肺纤维化过程中发挥了重要作用。另外,肥大细胞在硬皮病相关性肺病中的作用也引起了重视。SSc患者合并肺纤维化者肥大细胞比例以及组胺和胰酶水平与无肺纤维化者比起来是增高的。
总之,目前认为SSc肺间质纤维化的可能机制为,免疫及炎症效应细胞调节了肺组织的损伤及修复过程。免疫和炎症反应产生细胞因子和生长因子,通过调节成纤维细胞而影响了肺损伤和修复过程,促进了纤维化的发生。
SSc合并肺间质纤维化和IPF的病理表现很难鉴别。早期改变包括间质水肿,肺泡壁增厚,肺泡间隔内炎症细胞浸润(单核细胞、中性粒细胞、嗜酸性粒细胞和淋巴细胞等),结缔组织基质细胞浸润,成纤维细胞增生,胶原蛋白沉积,以及肺泡腔内炎症(主要是巨噬细胞),Ⅱ型肺泡细胞增生,不同程度的血管腔闭塞。病变主要分布在胸膜下、肺底以及肺后段。由于广泛的纤维化导致肺泡壁逐渐变薄、破裂,形成许多小气囊。还可能有肺动脉高压表现。SSc肺间质纤维化者病理改变以非特异性间质性肺炎类型为主,明显多于普通型间质性肺炎的类型。SSc合并间质性肺炎中,NSIP类型占77.5%,UIP类型占7.5%,终末期病变不能分类的有7.5%,其他类型包括呼吸性细支气管炎伴间质性肺病、结节病、机化性肺炎等。UIP病理学特点为病变在空间分布和时间分布上都存在明显的异质性:正常肺组织和病变组织间杂;不同部位的病变新旧程度不一,有些病灶处于活动性炎症期而另外一些则处于明显纤维化期,可以见到活跃增殖的成纤维细胞灶和胶原沉积或者蜂窝样改变同时存在。而NSIP特点为肺泡壁内有不同程度的炎症或者纤维化改变,但在时间上基本一致,无UIP新旧病灶共存的现象。电子显微镜可以显示早期的内皮细胞、表皮细胞损伤,在光镜无明显异常时即可出现上述改变。
1.血清学检查
90%SSc患者抗核抗体(ANA)呈阳性。特异性抗体为抗着丝点抗体(anticentromere antibody,ACA)和抗Scl-70抗体。抗Scl-70抗体又称抗拓扑异构酶Ⅰ(anti-topoisomeraseⅠ)抗体,更常见于弥漫性皮肤型SSc,与ILD发生风险增加相关;而ACA多见于局限性皮肤型SSc,与肺血管疾病和肺动脉高压关系更密切。
2.影像学表现
SSc患者X线胸片表现可为正常,或仅有轻度间质纹理增厚。典型的X线胸片表现为双肺对称的网状结节影,首先出现在双肺底及肺外带,随后病变逐渐往上发展,累及下2/3肺,但一般不累及肺尖。随着病情进展,网状结节影逐渐加重,甚至表现为蜂窝肺和肺容积缩小。少部分患者可有胸腔积液、胸膜增厚。胸腔积液的原因可能有胸膜受累,也可能是由于肺动脉高压或者心肌纤维化所致的充血性心力衰竭。和SLE或RA比较,SSc胸腔积液较少见。X线胸片还可以有肺动脉压增高的征象。
CT尤其是高分辨率CT(HRCT)对发现肺部受累敏感性更高,约91%SSc患者HRCT有肺间质改变。常见表现有磨玻璃样改变、不规则线状影、网状结节影、蜂窝样改变及牵引性支气管或细支气管扩张。病变首先出现在肺底、胸膜下及背侧,逐渐向中上肺、肺内侧及前侧发展(图1)。上述改变缺乏特异性,与SLE、RA及混合型结缔组织病的肺间质改变相似。
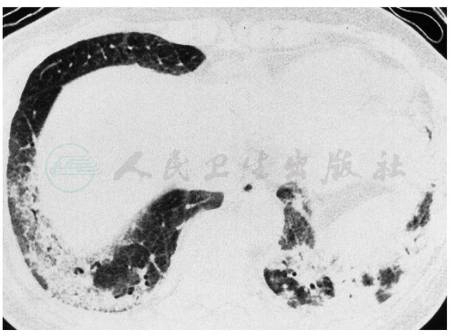
图1 SSc合并非特异间质肺炎
HRCT显示双肺基底部实变;肺活检证实为NSIP。
3.肺功能检查
肺功能改变为限制性通气功能障碍及弥散功能下降。限制性通气功能障碍表现为肺活量(VC)、肺总量(TLC)、残气量(RV)下降,肺顺应性降低。SSc患者的VC下降的速度是正常人群的3倍,BAL提示有活动性肺泡炎的患者VC下降更快。并且SSc发病的前2年内VC下降更快,故早期发现肺部病变具有重要意义。弥散功能(DL CO)下降是SSc肺间质病变最敏感的指标之一,对早期诊断有重要意义。较少见的改变有阻塞性或者混合型肺功能改变。运动肺功能检测显示:V/Q不匹配加重,PA-aO2增大,死腔通气增加、VD/VT增大。
4.血气分析
早期无明显改变。随着疾病进展可出现PaO2及SaO2下降,PaCO2正常或者下降。
5.支气管肺泡灌洗(BAL)
50%~60%SSc患者BAL有异常发现。在出现呼吸道症状或影像学改变前,BAL即可发现肺泡炎的存在,故有利于发现亚临床患者。BAL特点为肺泡巨噬细胞、中性粒细胞、嗜酸性粒细胞或者淋巴细胞增多,也可有IgG、FN、CIC等非细胞成分增多。通常,BAL中性粒细胞升高的患者CT往往表现为进展期病变、尤其是网格影改变(提示肺纤维化);而嗜酸性粒细胞增多与磨玻璃改变有关(提示肺泡炎)。BAL有肺泡炎表现者呼吸困难症状及影像学异常更明显,且肺功能恶化更快。BAL中性粒细胞升高者呼吸困难更明显、肺功能下降更快、预后更差,但也有研究并未观察到类似结果。虽然许多患者BAL结果正常,但病情却在进展,所以不宜单独用BAL的结果来指导治疗或者监测病情。
6.病理活检
经支气管肺活检(TBLB)受取材部位和标本量限制,对SSc-ILD的诊断价值有限。外科胸腔镜或者开胸肺活检是诊断ILD最可靠的标准。然而对于SSc外科肺活检并非必需,除非胸部HRCT不符合典型ILD,怀疑肉芽肿疾病或者其他疾病,与感染鉴别困难等情况下,可以考虑进行外科肺活检。
1.一般治疗
包括氧疗,肺康复治疗,控制心力衰竭及感染及治疗胃食管反流等。
2.药物治疗
肺泡炎持续存在者,肺容积及DLCO下降更明显、更快。故免疫抑制剂可用于治疗SSc所致的肺间质纤维化。环磷酰胺(CTX)合并低剂量泼尼松治疗可改善肺功能及预后。两者用于疾病早期可有效抑制肺泡炎。
CTD相关ILD中,SSc-ILD的随机对照研究数量有限。目前缺乏足够证据指导治疗,也无统一的治疗方案。治疗必须个体化,要充分衡量风险效益比。SSc-ILD患者以下情况可以考虑开始治疗:有呼吸道症状;有疾病活动或者进展证据,包括疾病处于相对较早阶段,肺功能异常并且有下降趋势,胸部HRCT有磨玻璃影等;并且无禁忌证,如感染、免疫缺陷、妊娠或哺乳等。
目前主要治疗药物是糖皮质激素和免疫抑制剂。常用的治疗方案为环磷酰胺和口服小剂量糖皮质激素(相当于泼尼松≤10mg/d)。不能使用环磷酰胺的患者,可以考虑吗替麦考酚酯(mycophenolate mofetil,MMF)或者硫唑嘌呤加小剂量糖皮质激素。
(1)糖皮质激素:
虽然在临床中糖皮质激素常用于治疗ILD,但是糖皮质激素对于SSc-ILD的疗效缺乏足够证据。糖皮质激素最佳剂量目前也没有统一意见。有专家认为泼尼松应≤10mg/d,并且应该联合其他免疫抑制剂,如环磷酰胺、吗替麦考酚酯治疗及硫唑嘌呤等。也有个别观察性研究显示,大剂量激素有短期疗效,但仍有待进一步研究。对于起病较急或者病情较重的患者,可以酌情给予泼尼松起始剂量0.5~1mg/(kg•d),逐渐减量。但是使用糖皮质激素需要慎重,一方面是缺乏足够的有效证据,有免疫抑制和感染风险,并且大剂量激素治疗对于SSc患者有导致硬皮病肾危象的风险。
(2)环磷酰胺:
环磷酰胺是治疗SSc-ILD最常用的免疫抑制剂。但是环磷酰胺治疗SSc-ILD目前仍存在争议。随机双盲安慰剂对照研究(scleroderma lung study)显示,158名有活动性肺泡炎的SSc-ILD患者口服环磷酰胺[≤2mg/(kg•d)]治疗12个月,可以减慢肺功能(FVC)下降速度,改善呼吸困难、活动耐力、健康相关生活质量和皮肤增厚。至24个月,除了呼吸困难有改善,环磷酰胺的其他作用不再显著。有荟萃分析研究了环磷酰胺对于SSc-ILD肺泡炎的疗效,结果显示环磷酰胺治疗12个月能够改善FVC恶化,但是不能够防止DLCO恶化。有临床意义的肺功能改善应大于10%,如果以此为标准,则环磷酰胺对于SSc-ILD的疗效并不显著。另有研究表明,环磷酰胺治疗ILD恶化的SSc患者,6个月和2年时分别有70%和51.8%的患者肺功能稳定或者改善。因此CTX对于进展或者加重期的SSc-ILD可能更有治疗价值,但仍需要进一步研究。总的来说,环磷酰胺可以用于SSc-ILD,但是剂量和疗程必须个体化,并且用药过程中应密切监测血常规、尿常规及肝肾功能。
(3)吗替麦考酚酯:
有数个回顾性研究及观察性队列研究显示,吗替麦考酚酯治疗SSc-ILD,患者肺功能改善或者稳定。另有前瞻性观察研究显示14名SSc-ILD患者接受吗替麦考酚酯治疗12个月,6例FVC改善大于10%,5例保持稳定。最新研究结果显示,与环磷酰胺对比,吗替麦考酚酯稳定或改善肺功能的疗效相似,但患者对吗替麦考酚酯的耐受性更好,副作用更小。
(4)硫唑嘌呤:
硫唑嘌呤治疗SSc-ILD的研究资料也很有限。小规模回顾性研究显示,11例有症状加重或者肺功能恶化的SSc-ILD患者,给予硫唑嘌呤和泼尼松治疗,8例治疗时间大于1年,其中5例FVC有改善,3例FVC稳定。另有研究比较硫唑嘌呤和环磷酰胺的疗效,结果环磷酰胺组FVC和DLCO稳定,而硫唑嘌呤治疗组FVC和DLCO恶化。也有研究显示患者完成CTX治疗后,可以用硫唑嘌呤维持治疗。
(5)自体干细胞移植:
另有研究比较了自体干细胞移植和静脉环磷酰胺治疗SSc。治疗12个月干细胞治疗组FVC显著改善,80%的患者改善持续至2年。这个结果提示干细胞移植可能比环磷酰胺有效,但仍有待进一步研究。
(6)利妥昔单抗(retuximab):
也有报道利妥昔单抗治疗环磷酰胺难治性SSc-ILD,结果显示症状、肺功能和HRCT有改善。
(7)抗纤维化治疗:
最近的研究结果显示尼达尼布可延缓患者肺功能的下降。
对于终末期肺间质纤维化患者,在无其他脏器活动性病变时可考虑行肺移植术。









