


 收藏
收藏 已收藏
已收藏英文名称 :lymphomatoid granulomatosis
淋巴瘤样肉芽肿病(lymphomatoid granulomatosis,LYG)是一种罕见的血管中心性和伴血管损伤的B细胞淋巴增生性疾病(LPD),常有B淋巴细胞增生紊乱,高度增生的T细胞浸润,并与EBV感染相关。临床上可多系统受累,肺受累最为常见(90%~100%),特征是多发性肺结节病灶,病理可见血管壁淋巴细胞浸润,也可以涉及皮肤、肾和神经系统。由Liebow等于1972年首先描述,当其在研究韦格纳(Wegener)肉芽肿时发现的一种淋巴结以外的以血管为中心伴血管损害的淋巴增生性病。开始因其兼有韦格纳肉芽肿和淋巴瘤的临床和病理学特征,难以确定是变异的韦格纳肉芽肿还是淋巴瘤,故称之为淋巴瘤样肉芽肿。目前认为LYG是由EBV阳性B淋巴细胞混合数量不等的反应性T细胞组成的血管中心和血管破坏性淋巴组织增生性疾病。2003年WHO肺肿瘤组织学分类将LYG列入淋巴组织增生性肿瘤项下AILⅡ级(恶性潜能未定)介于良性淋巴细胞血管炎及肉芽肿(AILⅠ级)与血管中心性淋巴瘤(AILⅢ级)之间。另外无论其组织学形态、侵蚀性还是疗效预后,LYG都具有良恶渐变的特点,部分已可诊断为B细胞淋巴瘤。在2008年和2016年版的WHO关于淋巴造血组织的肿瘤的分类中,把LYG归属为成熟B细胞肿瘤中弥漫性大B细胞淋巴瘤。
LYG至今病因不明,其发病与免疫功能抑制、先天性或后天性免疫功能不全有关,包括器官移植、自体干细胞移植、干燥综合征、HIV感染、X-淋巴增殖综合征和原发性免疫功能缺陷者患病风险均高于正常人。在几乎所有患者中,LYG似乎均与EBV感染有关。EB病毒感染机体后,可与B细胞结合,导致B细胞的单克隆性增生。正常机体通常可借助T细胞的免疫杀伤机制消灭病毒,但如患者出现免疫缺陷、免疫抑制或T细胞发育异常时,机体不能有效杀灭病毒并可导致感染的B细胞形成系列细胞因子,加重EBV的感染和促进病变细胞的增殖和转录,使其在T细胞和其他反应性细胞的伴随下无限制的克隆。
(一)大体表现
为灰黄或粉色结节,中心可有坏死和空洞形成。
(二)镜下表现
如图1所示,淋巴瘤样肉芽肿病病理形态具有以下特点:血管中心淋巴细胞浸润,细胞呈多样性,不同程度的坏死。病变有显著的血管中心和血管破坏性的分布特点。主要累及肌性动、静脉,血管壁全层有较多淋巴细胞浸润,内膜显著增厚,管壁狭窄,甚至闭塞,但管壁完整。除大片坏死区外,无灶状管壁的坏死和肌层的断裂,甚至在大片坏死区仅见残留病变血管结构。在早期或较小的病灶,病变主要局限于血管壁,随着病变扩大,可累及血管周围的肺组织。淋巴瘤样肉芽肿病浸润的细胞呈现多样性,有较多小淋巴细胞,少许组织细胞,浆细胞和数量不等体积较大的不典型淋巴细胞,细胞体积较大,核空泡状,可有双核或多核,核仁明显。但一般无中性粒细胞和嗜酸性粒细胞。尽管称其为淋巴瘤样肉芽肿病,但病变中无明显上皮样细胞肉芽肿和多核组织细胞。
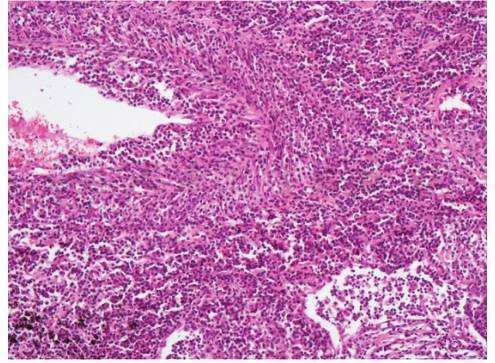
图1 肺组织内血管壁见较多淋巴细胞浸润
(三)免疫组织化学染色
如图2~图4所示,淋巴瘤样肉芽肿病的小淋巴细胞大部分为CD2、CD3、CD4、CD45RO阳性的B辅助淋巴细胞,少数为CD8阳性的T杀伤细胞和CD56阳性的自然杀伤细胞。不典型大淋巴细胞CD20、CD79a阳性的B细胞,部分病例显示轻链限制性和免疫球蛋白重链重排阳性,EBER(+)。
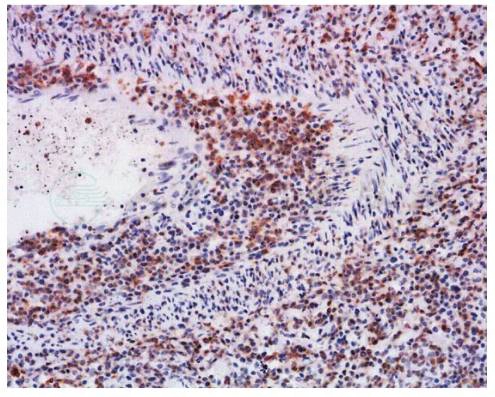
图2 CD3免疫组化染色显示较多小淋巴细胞阳性
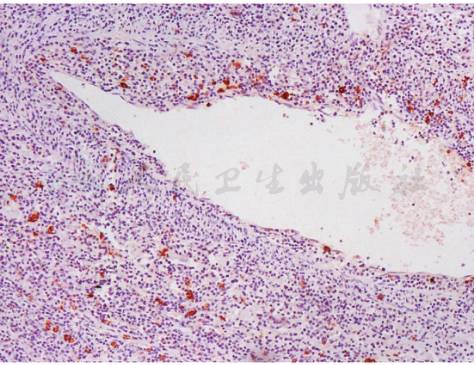
图3 CD20免疫组化显示散在大细胞阳性
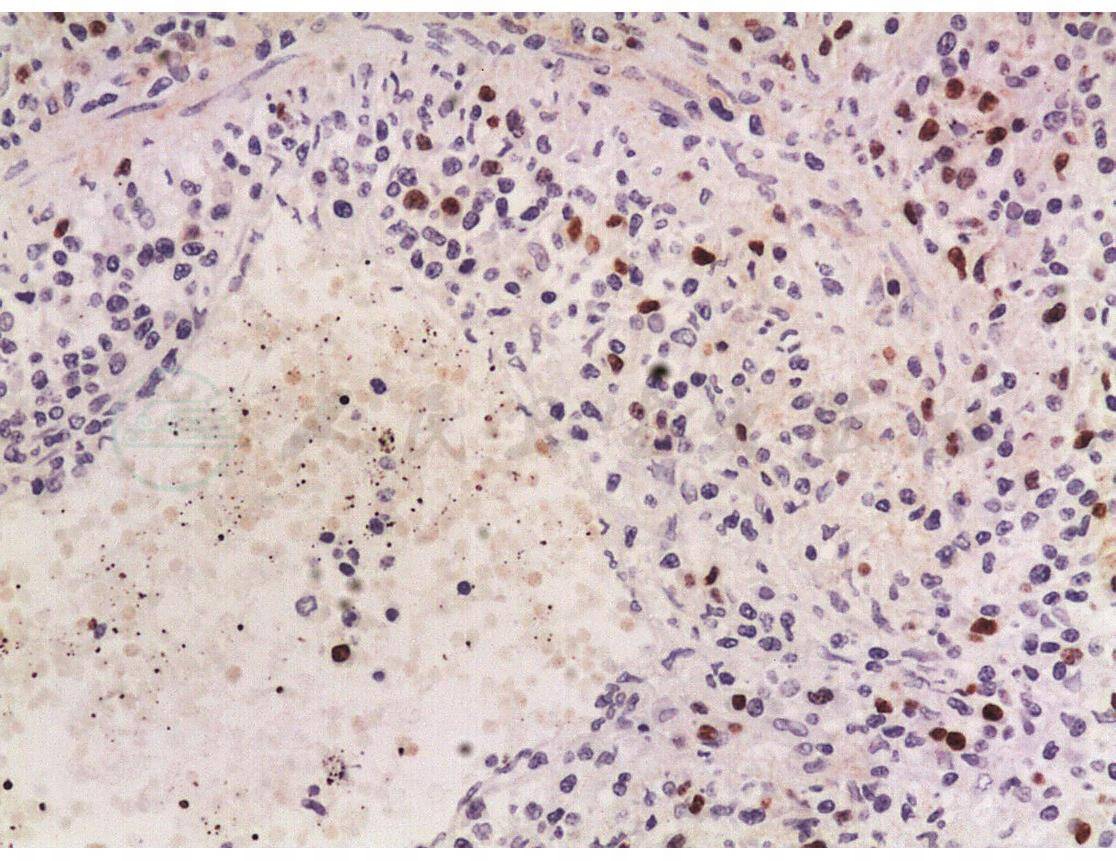
图4 EBV原位杂交散在阳性,EBER(+)
(四)组织分级
LYG的预后与其病变中的不典型淋巴细胞的数目有关,数量越多,预后越差。据此提出根据不典型淋巴细胞数量而定的3级分级系统,近来WHO分类对LYG组织分级提供了特殊标准,主要根据原位EBV阳性细胞数目和大B淋巴细胞的比例。
Ⅰ级:病变为血管中心性和细胞壁内淋巴细胞浸润,浸润细胞有小淋巴细胞、组织细胞,小淋巴细胞具轻微细胞异型性,大淋巴细胞及核分裂少见,小于1%。可见EBV RNA阳性细胞(约小于5个/HPF),无坏死或局部灶性坏死,呈良性病程。
Ⅱ级:病变浸润细胞为小淋巴细胞、多形性淋巴细胞,小淋巴细胞具不规则细胞核,可见大淋巴细胞,但少于细胞总数的5%。可见散在核分裂,有凝固性坏死,但不广泛。常见EBV RNA阳性细胞为5~20个/HPF,为交接病程。
Ⅲ级:病变体积明显增大,不典型大淋巴数量明显增多呈片状分布,小淋巴细胞有明显异型性和不规则的核,大淋巴细胞多,核染色质粗,可见明显核仁,核分裂易见,凝固性坏死广泛。许多细胞可见EBV RNA阳性(大于50个/HPF)。组织学分级越高,预后越差。Ⅲ级可视为弥漫大B细胞淋巴瘤的亚型,临床上可作为弥漫大B细胞淋巴瘤对待。
通过基因重组技术证实:大多数Ⅰ级病例为多克隆,而Ⅱ、Ⅲ级则多为单克隆免疫球蛋白。
(一)胸部X线及CT检查
是发现LYG的主要手段,但缺乏特异性,表现依病程而异,以双下肺周边多发的片状阴影、肿块影和结节影常见,沿支气管血管管束和小叶间隔分布。如图5所示,胸部CT显示病灶更清晰,能发现部分早期病变。根据形态不同,可分为4种不同类型:
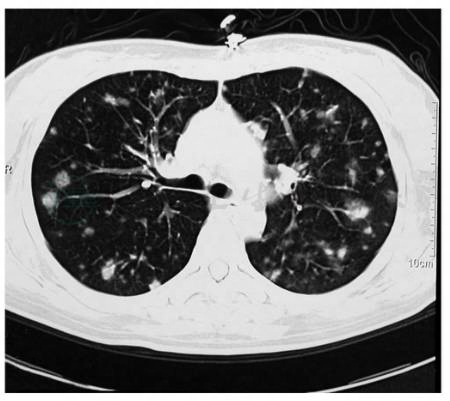
图5 胸部CT显示双肺多发结节影,沿支气管血管管束和小叶间隔分布
1.类肺炎型
表现为双肺大片状密度增高影,多位于两肺下野边缘模糊,病灶内可见支气管征象。
2.肿块型
表现为双肺多发大小不等的不规则肿块,边缘不光整、欠锐利,有分叶,无毛刺,可合并坏死、空洞。
3.结节型
表现为双肺多发大小不等的结节影,以中下肺野多见、结节边缘欠锐利。
4.混合型
表现为双肺大片状密度增高影及不规则肿块或大小不等的结节影。
(二)实验室检查
一般无特异性,部分患者白细胞计数增多,贫血,血沉正常或增快。肝酶轻度升高。类风湿因子可阳性,免疫球蛋白IgM或IgG轻度升高。
对肺原发性LYG迄今无标准的治疗方案。目前主要根据组织学分级来选择治疗手段。对于肺部病变较为局限的LYG患者,多主张积极手术切除或放射治疗,术后可行全身系统治疗。单用糖皮质激素治疗效果差,多种药物联合效果较好。通常以大剂量的糖皮质激素加环磷酰胺为基础的联合化疗报道最多。现认为组织病理学分级Ⅰ、Ⅱ级且临床无症状的患者推荐临床观察或糖皮质激素治疗,可选用泼尼松,常用剂量为1mg/kg,隔天1次。Ⅰ、Ⅱ级患者也可选择干扰素α-2b,用法1 000万U皮下注射,每周3次,持续40~60周。组织学Ⅰ、Ⅱ级但具有侵袭性的患者需单用或者联合化疗。强化治疗可用R-CHOP方案(利妥昔单抗、环磷酰胺、多柔比星、长春新碱、泼尼松)或者R-CVP方案(利妥昔单抗加环磷酰胺、长春新碱、泼尼松)。组织学Ⅲ级患者可按EBV阳性的大B细胞淋巴瘤治疗,一般可推荐用RCHOP或类似的强化治疗方案。在联合化疗失败者,骨髓移植可一定程度的缓解病情和延长生存期。
联合化疗无反应的患者还可尝试大剂量化疗加干细胞移植。近几年越来越多的靶向药物治疗研究作为传统化疗的补充手段。









