


 收藏
收藏 已收藏
已收藏英文名称 :mitochondrial myopathy
线粒体肌病(mitochondrial myopathy)和线粒体脑肌病(mitochondrial encephalomyopathy)是一组由线粒体DNA(mitochondrial DNA,mtDNA)或核DNA(nucleus DNA,nDNA)缺陷导致线粒体结构和功能障碍、ATP合成不足所致的多系统疾病,其共同特征为轻度活动后即感到极度疲乏无力,休息后好转;肌肉活检可见破碎红纤维(ragged red fiber,RRF)。如病变以侵犯骨骼肌为主,则称为线粒体肌病;如病变同时累及到中枢神经系统,则称为线粒体脑肌病。
线粒体肌病和线粒体脑肌病的病因主要是mtDNA(少数是nDNA)发生突变,如基因点突变(point mutation)、缺失(deletion)、重复(duplication)和丢失(depletion),即mtDNA拷贝数减少等,使编码线粒体在氧化代谢过程中所必需的酶或载体发生障碍,糖原和脂肪酸等原料不能进入线粒体或不能被充分利用,故不能产生足够的ATP。终因能量不足,不能维持细胞的正常生理功能,诱导细胞凋亡而导致线粒体病(mitochondrial disease)。
80%的线粒体脑肌病伴高乳酸血症和卒中样发作(mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes,MELAS)是由mtDNA第3243位点发生A到G的点突变(A3243G)所致。该突变由于改变了tRNA亮氨酸基因的结构,并进一步影响了线粒体蛋白质的合成和能量产生而致病。肌阵挛性癫痫伴破碎红纤维(myoclonus epilepsy ragged-red fibers,MERRF)主要是由于mtDNA第8344位点A到G的点突变(A8344G),使tRNA赖氨酸基因结构发生改变,蛋白合成受阻而致病。30%~50%的慢性进行性眼外肌瘫痪(chronic progressive external ophthalmoplegia,CPEO)和Kearns-Sayre综合征均有mtDNA的缺失,最常见缺失位于mtDNA的8468和13 446位之间。
1.肌肉
肌活检冷冻切片,经Gomori trichrome(GT)染色可见RRF(图1),由大量变性线粒体聚集造成。主要见于Ⅰ型肌纤维,油红O染色和糖原染色还可见脂肪和糖原堆积,肌组织内血管壁SDH染色阳性有助于诊断MELAS。电镜下可见肌膜下或肌原纤维间有大量异常线粒体,线粒体嵴排列紊乱,有时可见类结晶样包涵体(paracrystalline inclusions)。
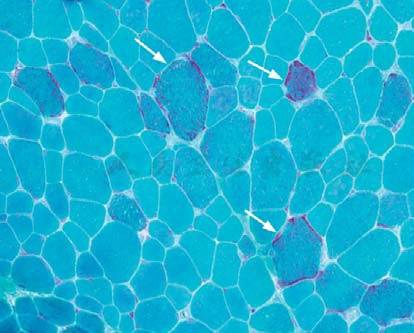
图1 线粒体肌病肌组织病理(GT×400)
肌纤维大小不等,可见RRF
2.脑
脑的病变复杂多样,广泛受累。主要为海绵样改变、神经元变性丢失、灶性坏死或广泛层性坏死、星形细胞增生、脱髓鞘或矿物质沉积。MELAS患者还可见颞顶枕叶皮质多灶性软化灶,脑皮质萎缩和基底核钙化,颅内多灶性坏死伴小血管增生和星形细胞增多,灶状或层状海绵样改变。MERRF患者可有齿状核、红核和苍白球等核团变性。
1.血生化检查
(1)乳酸、丙酮酸最小运动量试验:约80%的患者阳性,即运动后10分钟血乳酸和丙酮酸仍不能恢复正常。脑肌病者CSF乳酸含量也增高。
(2)线粒体呼吸链复合酶活性降低。
(3)约30%的患者的血清CK和LDH水平升高。
2.肌肉活检
见前面病理所述。
3.影像学检查
头颅CT或MRI示白质脑病、基底核钙化、脑软化、脑萎缩和脑室扩大。
4.肌电图
60%的患者为肌源性损害,少数呈神经源性损害或两者兼之。
5.线粒体DNA分析
对诊断有决定性意义。
(1)CPEO和KSS综合征均为mtDNA片段的缺失,其可能发生在卵子或胚胎形成的时期。
(2)80%的MELAS综合征患者是由于mtDNA tRNA亮氨酸基因位点3243的点突变所致。
(3)MERRF综合征主要是mtDNA tRNA赖氨酸基因位点8344的点突变所致。
目前无特效治疗,主要是对症治疗。主要的措施有:
1.饮食疗法
饮食治疗可减少内源性毒性代谢产物的产生。高蛋白、高碳水化合物、低脂饮食能代偿受损的糖异生和减少脂肪的分解。
2.药物治疗
可给予静脉滴注ATP 80~120mg及辅酶A 100~200U,每日一次,持续10~20天,以后改为口服ATP。辅酶Q10和大量B族维生素可使血乳酸和丙酮酸水平降低。左卡尼汀可以促进脂类代谢、改善能量代谢,成人1~3g/d,分2~3次口服,儿童50~100mg/(kg·d),每日最大剂量不超过3g。若血清肌酶谱明显升高可选择皮质激素治疗。对癫痫发作、颅压增高、心脏病、糖尿病等进行对症治疗。另外,中药如黄芪、党参、枸杞子等补气活血治疗及综合调理也可改善症状。
3.其他
物理治疗可减轻痛苦。KSS患者重度心脏传导阻滞者可用心脏起搏器。最根本的治疗有待于正在研究的基因治疗。









