


 收藏
收藏 已收藏
已收藏英文名称 :progressive muscular dystrophy
进行性肌营养不良症(progressive muscular dystrophy,PMD)是一组遗传性肌肉变性疾病,临床特征主要为缓慢进行性加重的对称性肌肉无力和萎缩,无感觉障碍。遗传方式主要为常染色体显性、隐性和X连锁隐性遗传。电生理表现主要为肌源性损害、神经传导速度正常。组织学特征主要为进行性的肌纤维坏死、再生和脂肪及纤维结缔组织增生,肌肉无异常代谢产物堆积。治疗方面主要为对症治疗,目前尚无有效的根治方法。
根据遗传方式、起病年龄、萎缩肌肉的分布、病程进展速度和预后,进行性肌营养不良症至少可以 分为9种类型:假肥大型肌营养不良症(pseudohypertrophy muscular dystrophy),包括Duchenne型肌营养不良症(Duchenne muscular dystrophy,DMD)和Becker型肌营养不良症(Becker muscular dystrophy,BMD)、面肩肱型肌营养不良症(facioscapulohumeral muscular dystrophy,FSHD)、肢带型肌营养不良症(limb-girdle muscular dystrophy,LGMD)、Emery-Dreifuss肌营养不良症(Emery-Dreifuss muscular dystrophy,EDMD)、先天性肌营养不良症(congenital muscular dystrophy,CMD)、眼咽型肌营养不良症(oculopharyngeal muscular dystrophy,OPMD)、眼肌型肌营养不良症(ocular muscular dystrophy)和远端型肌营养不良症(distal muscular dystrophy)。在这些类型中,DMD最常见,其次为BMD、FSHD和LGMD。
进行性肌营养不良症的各种类型的基因位置、突变类型和遗传方式均不相同,其致病机制也不一样。实际上各种类型均是一种独立的遗传病。如假肥大型肌营养不良症(DMD和BMD)的基因位于染色体Xp21,属X连锁隐性遗传。该基因全长约2400kb,是迄今为止发现的人类最大基因,cDNA长14kb,含79个外显子,编码3685个氨基酸,组成427KD的细胞骨架蛋白—抗肌萎缩蛋白(dystrophin)。该蛋白主要位于骨骼肌和心肌细胞膜的质膜面,具有细胞支架、抗牵拉、防止肌细胞膜在收缩活动时撕裂的功能。作为细胞骨架的主要成分,抗肌萎缩蛋白与肌纤维膜上的多种糖蛋白结合为抗肌萎缩蛋白相关蛋白复合体(dystrophin-associated protein complex,DAPC),这些复合体可与基膜层粘连蛋白(laminin)连接,以维持肌纤维的稳定性。DMD患者因基因缺陷而使肌细胞内缺乏抗肌萎缩蛋白,造成肌细胞膜不稳定并导致肌细胞坏死和功能缺失而发病。DMD患者大脑皮质神经元突触区抗肌萎缩蛋白的缺乏可能是智力发育迟滞的原因。
FSHD基因定位在4号染色体长臂末端(4q35),在此区域有一与KpnI酶切位点相关的3.3kb重复片段。正常人该3.3kb/KpnI片段重复10~150次,而FSHD患者通常少于8次,故通过测定3.3kb/ KpnI片段重复的次数则可作出基因诊断。FSHD患者3.3kb/KpnI片段重复次数的减少并不直接引起基因的结构破坏,而是引起4q35基因的转录抑制被减弱或消除,使其表达上调而致病。
肢带型肌营养不良症是一类具有高度遗传异质性和表型异质性的常染色体遗传性肌病。根据遗传方式,常染色体显性遗传的称为LGMD1,常染色体隐性遗传的称为LGMD2。各自按每一个不同的致病基因分为不同的亚型,如LGMD1分为LGMD1A、1B、1C、1D和1E五个类型;LGMD2分为LGMD2A、2B、2C、2D、2E、2F、2G、2H、2I和2J十个类型。90%以上的肢带型肌营养不良症是常染色体隐性遗传,以LGMD2A型最常见。肢带型肌营养不良的发病与肌膜蛋白和近膜蛋白的异常有关,直接影响肌细胞膜上的抗肌萎缩蛋白-糖蛋白复合体的结构和功能。复合体内各蛋白之间紧密结合,互相关联,作用为连接膜内骨架蛋白和膜外基质以保持肌细胞膜的稳定性。任何一种蛋白的缺失均会影响到整个膜结构的稳定,导致肌细胞的坏死。
眼咽型肌营养不良症基因位于染色体14q11.2-13,其蛋白产物为多聚腺苷酸结合蛋白2(polyadenylate-binding protein 2,PABP2),故也称多聚腺苷酸结合蛋白2基因。PABP2蛋白存在于细胞核中,对信使RNA起增加poly(A)的作用。发病机制与PABP2基因1号外显子上的GCG重复突变增加有关:正常人仅6次重复,而眼咽型肌营养不良症患者GCG重复8~13次,编码异常的多聚丙氨酸链。重复的次数越多,症状越重。
Emery-Dreifuss肌营养不良症基因位于染色体Xq28和1q21-23,分别编码emerin和核纤层蛋白A/C(laminA/C),主要位于骨骼肌、心肌、平滑肌核膜。该基因异常导致核膜稳定性受损,造成骨骼肌和心肌的损害。
各种类型的进行性肌营养不良症的肌肉病理改变主要为肌纤维的变性、坏死、萎缩和再生,肌膜核内移增多。随着病情进展,光镜下肌细胞大小差异不断增加,有的萎缩,有的代偿性增大,呈镶嵌分布;萎缩的肌纤维间有大量的脂肪细胞和结缔组织增生。Ⅰ型和Ⅱ型肌纤维均受累,为非特异性改变(图1)。电镜下肌原纤维排列紊乱或断裂,Z线破坏或消失,肌细胞膜有锯齿状改变。各种类型的特异性蛋白改变需用相应的抗体进行检测,如DMD和EDMD患者的肌活检标本分别用抗肌萎缩蛋白抗体和emerin抗体进行免疫组化染色可见抗肌萎缩蛋白和emerin蛋白缺失,对诊断有决定性意义。
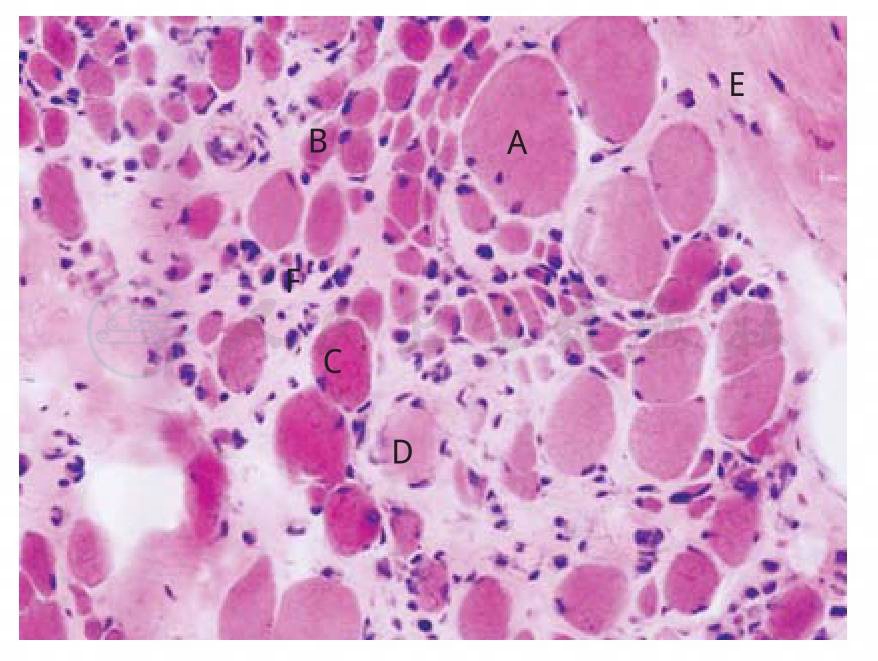
图1 DMD肌组织病理(HE×400)
肌纤维大小不等,可见肌纤维肥大(A)、萎缩(B)、变性(C)和坏死(D),肌束膜(E)和肌内膜(F)结缔组织明显增生
1.血清酶学检测
常规的血清酶检测主要包括肌酸激酶(creatine kinase,CK)、乳酸脱氢酶(lactate dehydrogenase,LDH)和肌酸激酶同工酶(creatine kinase-MB,CK-MB)。异常显著升高(正常值的20~100倍)者见于DMD、BMD、远端型肌营养不良症的Miyoshi亚型和LGMD2C、2D、2E、2F型。其他类型的肌酶轻到中度升高。在DMD和LGMD2晚期,因患者肌肉严重萎缩则血清CK值可明显下降。其他血清酶如谷氨酸草酰乙酸转氨酶(glutamic oxaloacetic transaminase,GOT)、谷氨酸丙酮酸转氨酶(glutamate-pyruvate transaminase,GPT)等在进展期均可轻度升高。
2.肌电图
具有典型的肌源性受损的表现。用针电极检查股四头肌或三角肌,静息时可见纤颤波和正锐波;轻收缩时可见运动单位时限缩短,波幅减低,多相波增多;大力收缩时可见强直样放电及病理干扰相。神经传导速度正常。
3.基因检查
采用PCR、MLPA、印迹杂交、DNA测序等方法,可以发现基因突变进行基因诊断。如用多重PCR或MLPA法可检测DMD基因外显子的缺失;印迹杂交法可进行FSHD基因诊断;DNA测序可明确LGMD等基因的突变碱基。
4.肌肉活检
大多数类型的进行性肌营养不良症患者的肌肉活检均表现为肌肉的坏死和再生、间质脂肪和结缔组织增生这一共性,常规染色方法不能区分各种类型,但采用免疫组织化学法使用特异性抗体可以检测肌细胞中特定蛋白是否存在,以此来鉴别各种类型的肌营养不良症。如用抗肌萎缩蛋白抗体检测DMD和BMD、用γ-肌聚糖蛋白(γ-sarcoglycan)抗体检测LGMD2C、用α-肌聚糖蛋白抗体检测LGMD2D、用β-肌聚糖蛋白抗体检测LGMD2E和用Emerin蛋白抗体检测EDMD等。
5.其他检查
X线、心电图、超声心动图可早期发现进行性肌营养不良症患者的心脏受累的程度。CT可发现骨骼肌受损的范围,MRI可见变性肌肉呈不同程度的“蚕食现象”。DMD和BMD患者应做智力检测。
进行性肌营养不良症迄今无特异性治疗,只能对症治疗及支持治疗,如增加营养,适当锻炼。物理疗法和矫形治疗可预防及改善脊柱畸形和关节挛缩,尤其是早期进行踝关节挛缩的矫正,对维持行走功能很重要。应鼓励患者尽可能从事日常活动,避免长期卧床。药物可选用ATP、肌苷、维生素E、肌生注射液和补中益气的通塞脉片等。基因治疗(外显子跳跃、微小基因替代)及干细胞移植治疗有望成为有效的治疗方法。
由于目前尚无有效的治疗方法,因此检出携带者、进行产前诊断、人工流产患病胎儿就显得尤其重要。首先,应确定先症者(患儿)的基因型,然后确定其母亲是否是携带者。当携带者怀孕以后确定是男胎还是女胎,对男胎进行产前基因诊断,若是病胎则终止妊娠,防止患儿出生。









