


 收藏
收藏 已收藏
已收藏英文名称 :inherited protein S deficiency
PS基因位于3号染色体。两种PS基因:PSβ基因为静止基因,PSα基因为表达基因。PSα基因全长80kb,含15个外显子、14个内含子,其cDNA已克隆。PS基因只有一个酶切位点,编码脯氨酸(Pro)626基因有两种自然型CCA/ CCG(发生率0. 52/0. 48),另4种已有报道的变异为PS Hearlen缺陷是丝氨酸460→脯氨酸,患者常不伴血栓病;PSβ基因天冬酰胺458(Asn458)变异使MspI酶切位点丢失,伴致血栓倾向。PSα基因外显子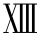 5. 3基因缺失产生Ⅰ型PS缺乏症,另一报道PS编码序列中部的基因缺失产生Ⅰ型PS缺乏症。
5. 3基因缺失产生Ⅰ型PS缺乏症,另一报道PS编码序列中部的基因缺失产生Ⅰ型PS缺乏症。
PS缺乏症是一种常染色体显性遗传性疾病,但有18%患者无家族史。自1984年文献上陆续报道以来,尚未知其确切发生率。估计PS缺乏症的发生率为1/15 000~1/20 000。在静脉血栓人群中的患病率占2%~13%,平均发病年龄28岁。
PS缺乏症分三型:见表1。三种类型的PS缺乏症的命名尚未有统一意见。按1991年ISTH会议建议分为Ⅰ、Ⅱa、Ⅱb型。Comp等于1984年、1986年命名为Ⅰ型者目前属于Ⅱ型PS缺乏症。
表1 遗传性PS缺乏症分型
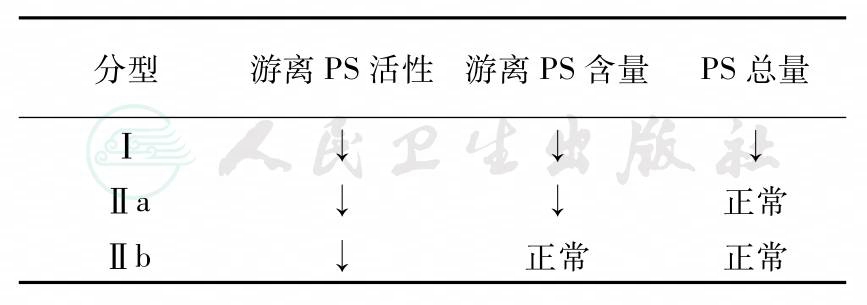
Ⅰ型PS缺乏症为经典型,结合及游离型PS都减少(<50%正常值)。Ⅱ型PS缺乏症为PS分子功能缺陷或激活型与游离型分布异常。后者使游离PS:Ag与活性都降低(<40%正常值)且不成比例。以Ⅱa型多见。Ⅰ型分子缺陷基础可能是基因缺失、点突变引起错义、误义或碱基对插入、并接区异常等。Ⅱ型的分子缺陷性质未明,可能由PS或C4bP基因变异致PS分布或功能异常所致。口服抗凝患者由于维生素K拮抗剂本身能降低PS水平。PS与凝血酶原或FⅩ比值降低,则有诊断价值。先天性PS缺乏症的诊断需与获得性PS缺乏症相鉴别。
①总PS测定:正常血浆总PS:Ag为23μg/ml(成人,RIA或ELISA测定法),若用Laurell火箭电泳法检测则正常值为72%~148%;②游离PS:Ag测定:由PEG 6000,4℃下将PS- C4b复合物沉淀,然后再以免疫法测定PS;或采用交叉免疫电泳,先将患者枸橼酸抗凝血浆4℃电泳,然后加入羊抗PSIgG在25℃交叉电泳,因PS泳动较复合物快,出现二个峰;③游离PS:C测定,由于测定PS为APC抗凝活性提供辅因子,在测定系统中需加入PS缺乏血浆。即患者血浆先和PS缺乏血浆按一定比例混合,然后加入蛇毒活化蛋白C并测定凝血时间。由于PS缺乏血浆制备困难,PS功能测定尚缺乏试剂盒。PS:C不能用发色底物法,因PS不能转化成丝氨酸蛋白酶。
血栓栓塞急性期采用肝素抗凝或溶栓治疗,口服抗凝剂用于预防血栓复发。合成雄激素(danazol)对PS缺乏症无效。









