


 收藏
收藏 已收藏
已收藏
基本信息
英文名称 :gangliosidosis
中文别名 :Gaucher病
概述
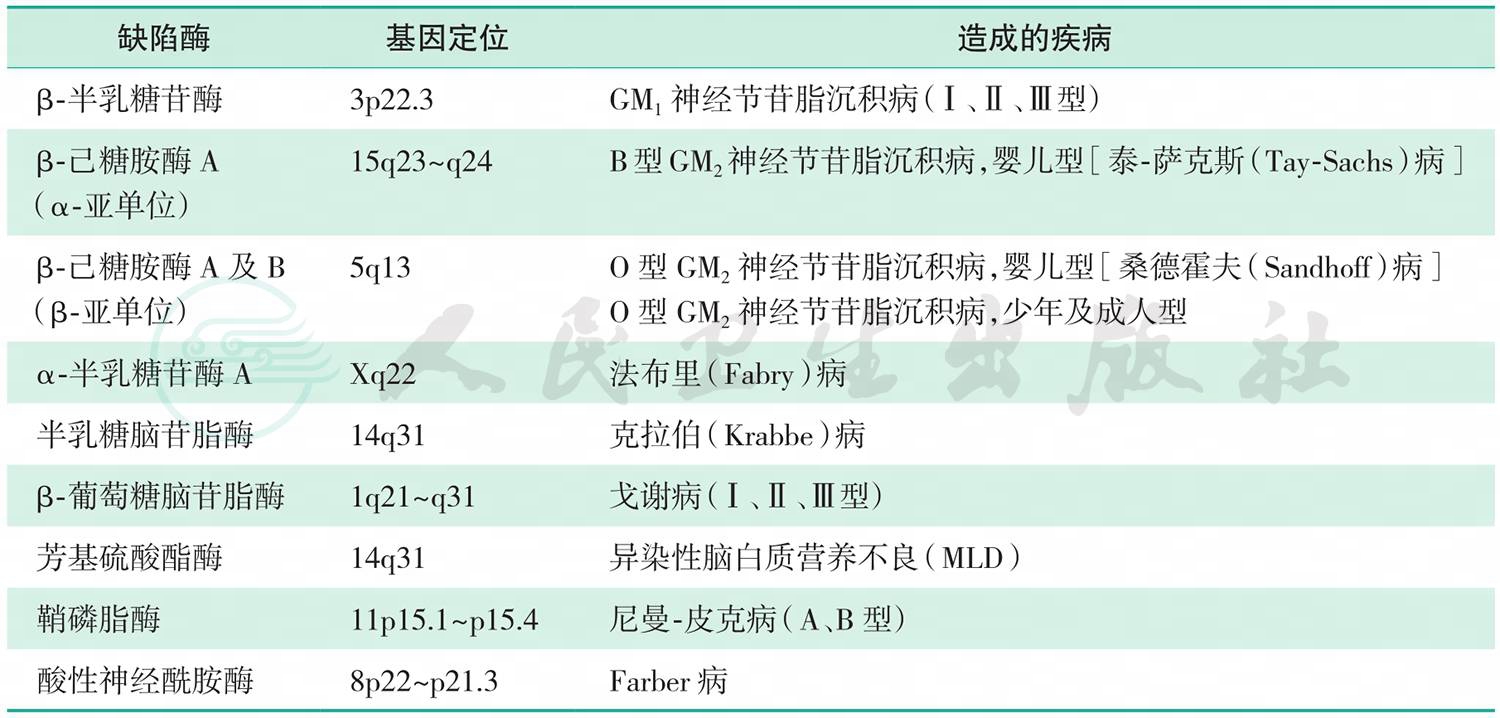
神经节苷脂广泛存在于人体各种细胞内,以脑和神经组织中含量最高。GM1是最主要的一种神经节苷脂,GM1的降解必须在溶酶体中经一系列水解酶的作用逐步进行,其中任一酶的缺陷都将造成节苷脂在溶酶体中沉积,进而破坏细胞和器官,导致神经节苷脂沉积病(gangliosidosis),其临床表现以中枢神经系统症状为主。溶酶体酶缺陷导致的各种脂质沉积症见表1。
表1溶酶体酶缺陷导致的各种脂质沉积症
病因学
GM1神经节苷脂沉积病是由酸性β-半乳糖苷酶(acid β-galactosidase)缺乏,阻断了GM1降解过程所造成。该酶编码基因位于3p22.3。
临床表现
此内容为收费内容
诊断
此内容为收费内容
鉴别诊断
此内容为收费内容
治疗
1.GM1神经节苷脂沉积病
迄今尚无有效治疗方法,预后不良,发病越早进展越快。通过抗惊厥药物等对症治疗及营养支持,部分患儿症状可暂时缓解,随着神经系统损害的进展出现吞咽困难、呛咳,反复呼吸道感染,多数患儿死于肺炎等并发症。骨髓移植、基因治疗、底物消除治疗等尚在研究中。
2.GM2节苷脂沉积病
本病无特殊治疗方法,主要是对症治疗。酶替代疗法、骨髓移植及基因疗法正在试用阶段。
3.GM3神经节苷脂病
多在婴儿期死于肺炎。
预防
1.GM1神经节苷脂沉积病
通过胎盘绒毛细胞、羊水细胞β-半乳糖苷酶活性测定,可进行本病的产前诊断,减少患儿的出生。通过婚前基因筛查,避免杂合子婚配,可有效地降低发病率。
2.GM2节苷脂沉积病
产前诊断主要依赖羊水细胞酶活性测定。尽早发现,及时终止妊娠。
作者
杨艳玲
来源









