


 收藏
收藏 已收藏
已收藏英文名称 :homocystinemia
中文别名 :同型半胱氨酸尿症
同型半胱氨酸血症(homocystinemia)又称同型半胱氨酸尿症(homocystinuria),是相对常见的可治疗的氨基酸代谢病,为常染色体隐性遗传病。
同型半胱氨酸大部分通过两条途径进行再甲基化、恢复成甲硫氨酸。其中一条途径是由甜菜碱提供甲基,由甜菜碱-同型半胱氨酸甲基转移酶催化,另一条途径是由甲基四氢叶酸提供甲基,经5-甲基四氢叶酸-同型半胱氨酸甲基转移酶催化进行,这一过程尚需维生素B12的衍生物甲钴铵作为辅助因子参与。因此,维生素B12代谢异常也可导致这一途径发生障碍。
已知的甲硫氨酸代谢途径中的酶缺陷有9种,经典型同型半胱氨酸血症共3型(表1),有些涉及钴胺素(维生素B12)的遗传性代谢缺陷。胱硫醚合酶(cystathionine synthase,CBS)缺乏症导致的同型半胱氨酸血症Ⅰ型是最严重的类型。CB S基因位于染色体21q22.3,含23个外显子,已发现160余种突变类型,有报道p.Gly307Ser等位基因突变预测维生素B6无反应,而p.Ile278 Thr等位基因变异通常对维生素B6有反应,CBS基因其他变异与维生素B6反应性无相关性。
表1 经典型同型胱氨酸血症三型的病因、临床、生化及遗传特点
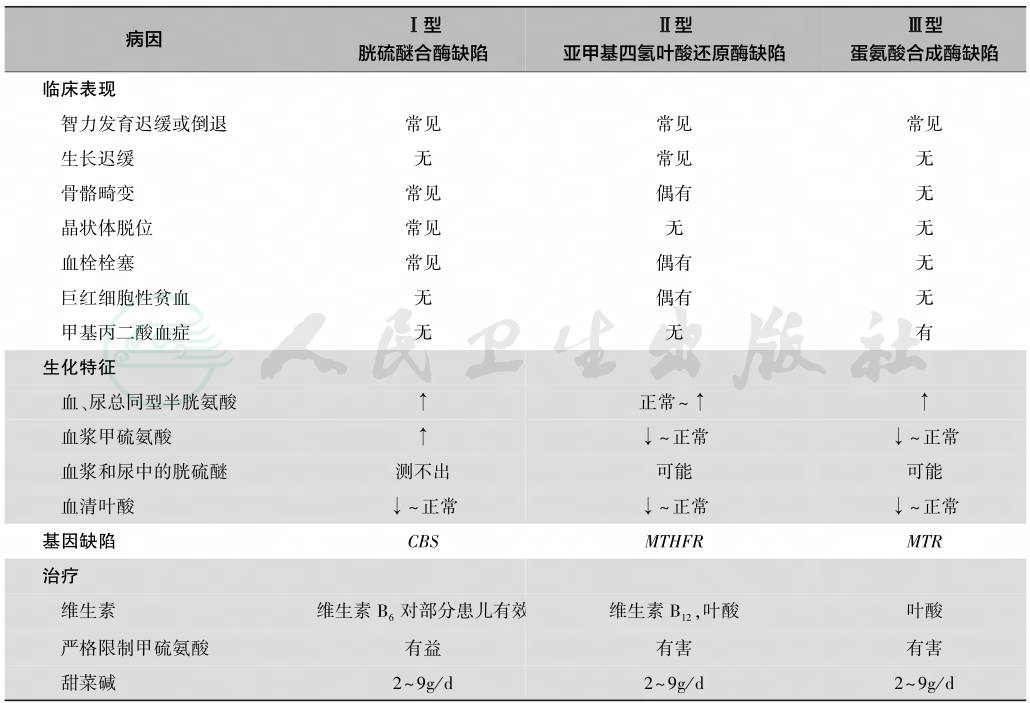
同型半胱氨酸血症Ⅱ型病因为编码亚甲基四氢叶酸还原酶的MTHFR基因致病变异,MTHFR位于染色体1p36.22,含11个外显子。欧洲、亚洲、美洲、中东和澳大利亚MTHFR基因c.677C>T多态性研究发现,TT基因型在中国北方(20%)、意大利南部(26%)和墨西哥(32%)尤为常见,是引起高血压、心脑血管疾病的主要原因之一,一些患儿易发生神经精神损害。
同型半胱氨酸血症Ⅲ型病因为编码蛋氨酸合成酶(methionine synthase,MS)的基因 MTR 缺陷,MTR 位于染色体1q43,含33个外显子,以错义突变常见。
1.新生儿筛查或高危筛查
血液蛋氨酸浓度增高,总同型半胱氨酸增高,胱硫醚和胱氨酸下降,为同型半胱氨酸血症Ⅰ型的特征。
2.尿硝普盐试验
可作为疑诊患儿的初筛方法,尿液中含有同型(半)胱氨酸、胱氨酸时亦呈阳性结果。
3.尿液检查
总同型半胱氨酸显著增高,有机酸正常。
4.酶学检测
同型半胱氨酸血症Ⅰ型患儿淋巴细胞、皮肤成纤维细胞、肝、脑、胰等组织胱硫醚合成酶活性降低。
5.基因诊断
相关基因双等位致病变异。
1.药物及饮食治疗
(1)维生素B6
对约半数同型半胱氨酸血症Ⅰ型患儿有效,剂量因人而异,100~1 000mg/d,同时应加用叶酸或亚叶酸5~10mg/d;当每日口服500~1 000mg数周而血生化指标无好转时,可视为维生素B6无反应型。
(2)低蛋氨酸-高胱氨酸饮食
对部分同型半胱氨酸血症Ⅰ型患儿有效,需限制天然蛋白质,补充无蛋氨酸的特殊治疗用配方奶粉。对于同型半胱氨酸血症Ⅱ、Ⅲ型患儿,无需限制蛋白质,应正常饮食。
(3)甜菜碱
用于非维生素B6反应型同型半胱氨酸血症Ⅰ型患儿的治疗,每日2~9g,分次服用。有助于改善同型半胱氨酸血症Ⅱ、Ⅲ型患儿的临床症状。
治疗过程中应定期监测生长速率、神经精神及骨骼情况、血和尿的氨基酸,维持血浆蛋氨酸浓度<40μmol/L;血和尿中的总同型半胱氨酸浓度应维持在正常范围。
2.肝移植
对于饮食及药物治疗控制不良的同型半胱氨酸血症Ⅰ型患儿,可考虑肝移植。
新生儿筛查是发现同型半胱氨酸血症Ⅰ型的重要措施,如果在发病后开始治疗,患儿可能遗留不可逆性脑损害。如能在症状前开始治疗,部分患儿可以获得正常发育。
对于基因诊断明确的家系,可在母亲下一次妊娠8~13周左右留取胎盘绒毛,或在妊娠16~22周抽取羊水,分取羊水细胞,通过胎儿基因分析进行产前诊断。









