


 收藏
收藏 已收藏
已收藏英文名称 :tetrahydrobiopterin deficiency
四氢生物蝶呤(tetrahydrobiopterin deficiency,BH4)缺乏症为常染色体隐性遗传病,因其合成或代谢途径中某种酶的先天性缺陷导致一些芳香族氨基酸代谢障碍,影响脑内神经递质合成,患儿出现严重的神经系统损害症状和智能障碍。常见的BH4缺乏症包括6-丙酮酰四氢蝶呤合成酶(6-pyruvoyl tetrahydropterin synthase,PTPS)缺乏症及二氢蝶啶还原酶(dihydropteridine reductase,DHPR)缺乏症,少见为鸟苷三磷酸环化水解酶(guanosine triphosphate cyclohydrolase,GTPCH)缺乏症、蝶呤-4α-二甲醇胺脱水酶(pterin 4α-carbinolamine dehydrogenase,PCD)缺乏症及墨蝶呤还原酶缺乏症。
BH4缺乏症在高苯丙氨酸血症(HPA)中的比例各国家不一,白种人约占1%~2%;日本占4%,韩国10%。我国已诊断BH4缺乏症近300例,南方地区发生率高于北方,总体在高苯丙氨酸血症中约占10%~15%,以PTPS缺乏症最常见。
BH4是苯丙氨酸羟化酶、酪氨酸羟化酶、色氨酸羟化酶的辅酶。BH4合成和代谢途径见图1。BH4与Phe通过一种GTP环化水解酶Ⅰ反馈调节蛋白(GFRP)起着调节GTPCH作用,BH4负反馈抑制GTPCH,苯丙氨酸(Phe)可增加GFRP作用,因此当血Phe增高时,通过增强GTPCH作用使新蝶呤和生物蝶呤的合成也相应增高,通过三磷酸鸟苷(GTP)在GTPCH、PTPS和SR三种合成酶作用下合成增加,再经PCD作用后生成醌-二氢生物蝶啶(BH2),在二氢生物蝶啶还原酶(DHPR)作用下生成具有生物活性的BH4,发挥重要的生理作用。BH4代谢途径中任何一种合成酶或还原酶缺乏均可导致BH4生成不足或完全缺乏。
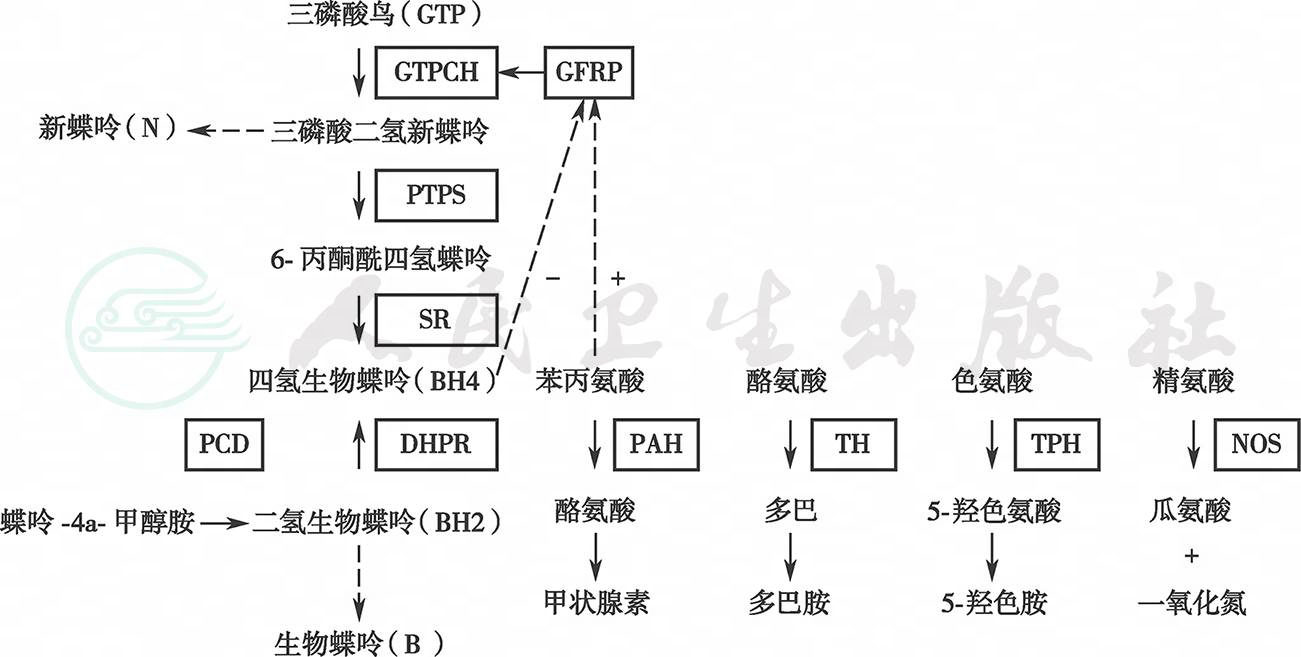
图1四氢生物蝶呤合成代谢示意图
GTPCH:GTP环化水解酶;PTPS:6-丙酮酰四氢蝶呤合成酶;SR:墨蝶呤还原酶;PCD:蝶呤-4α-甲醇胺脱水酶;DHPR:二氢蝶啶还原酶;PAH:苯丙氨酸羟化酶;TH:酪氨酸羟化酶;TPH:色氨酸羟化酶;NOS:一氧化氮合成酶
引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
BH4缺乏不仅影响苯丙氨酸羟化酶的稳定性,使酶活性下降,导致血Phe浓度增高,出现类似与经典型PKU的代谢异常,而且降低了酪氨酸羟化酶、色氨酸羟化酶活性,导致神经递质前质左旋多巴胺(L-DOPA)和5-羟色氨酸生成受阻,从而影响了脑内神经递质多巴胺、5-羟色胺的合成,使患者出现严重的神经系统损害的症状和体征,预后比经典型PKU更差。
DHPR基因QDPR位于4p15.3,含7个外显子,编码蛋白为244个氨基酸。GTPCH基因GCH1,位于14q22.1-q22.2,含6个外显子,编码222氨基酸。PTPS的基因PTS,位于11q22.3,全长2kb,有6个外显子。根据我国143例BH4D患者的基因突变类型分析,发现PTS基因有32种突变,其中c.155A>G,c.259C>T,c.286G>A和c.IVS1-291A>G为热点突变(占76.9%),c.259C>T在中国南北方都多见,c.155A>G则多见于南方患者;c.155A>G,c.259C>T,c.286G>A可导致严重型PTPS缺乏有关,c.166G>A(V56M)及c.IVS1-291A>G可能与轻型PTPS缺乏症有关。至今中国已发现8例DHPR缺乏症,发现10种QDPR基因突变。
(一)实验室检查
1.新生儿筛查
出生48小时后采集干滤纸血片进行血Phe浓度测定,对所有高苯丙氨酸血症患者进行尿蝶呤谱分析及血DHPR活性测定进行BH4缺乏症鉴别。BH4缺乏症进行早期治疗,可避免神经系统损害和智能障碍发生。BH4缺乏症患者血Phe增高程度变异大,可轻度增高,或类似经典型PKU。
2.尿蝶呤谱分析
对BH4合成酶缺乏(PTPS及GTPCH缺乏症)诊断较可靠。新鲜尿液收集后马上加入抗坏血酸(每ml 尿液加10~20mg抗坏血酸),避光下混合均匀后-70℃保存或浸透5cm×5cm大小专用滤纸片上,避光晾干后邮寄检测。实验室采用高效液相仪(HPLC)进行尿新蝶呤(neopterin,N)、生物蝶呤(biopterin,B)定量分析,从而得出两者之比例和生物蝶呤百分率[B/(B+N)×100%]。
PTPS缺乏时,尿新蝶呤(N)明显增加,生物蝶呤(B)明显降低,B% < 10%(多数 < 5%);对于尿新蝶呤明显增高,尿生物蝶呤正常或略低,B%介于5%~10%,诊断需谨慎,可结合BH4负荷试验协助诊断。还原酶DHPR缺乏时,尿新蝶呤可正常或稍高,生物蝶呤明显增加,B%增高,但部分DHPR缺乏患者可有正常尿蝶呤谱;GTPCH缺乏者,尿新蝶呤、生物蝶呤均极低,B%正常;PCD缺乏者在生物蝶呤峰后出现7-生物蝶呤波峰(需要有特异内标);SR缺乏症尿蝶呤谱可正常。
3.二氢蝶啶还原酶分析
红细胞二氢蝶啶还原酶活性测定是DHPR缺乏症的确诊方法。由于常规尿蝶呤谱分析和BH4负荷试验并不能完全对DHPR缺乏症进行鉴别,需要通过红细胞DHPR活性测定以确诊。分析采用双光束分光光度计,测定外周血干血滤纸片中的DHPR活性。
4.BH4负荷试验
BH4负荷试验是BH4缺乏症辅助诊断试验和鉴别BH4反应性PKU/HPA的有效方法。常采用人工合成BH4药物(二盐酸沙丙蝶呤,Kuvan)(每片100mg)进行负荷试验。试验前先留尿做尿蝶呤谱分析,血Phe>600µmol/L(新生儿>400µmol/L)的患儿可在喂奶前30分钟给予口服BH4片(20mg/kg),BH4 服前,服后2、4、6、8,24小时分别取血作Phe、Try测定,服后4~8小时留尿做尿蝶呤谱分析。PTPS缺乏者,当给予BH4后,因其苯丙氨酸羟化酶活性恢复,血Phe浓度多在服用BH4后4~6小时下降80%~90%或降至正常;DHPR缺乏者血Phe下降缓慢,类似部分BH4反应型PKU/HPA。
(二)影像学检查
BH4缺乏症MRI检查,T1加权成像可发现在豆状核对称性钙化灶、脑沟脑回深,皮质下囊性变,T2加权成像显示脱髓鞘病变导致的脑室周围脑白质高信号改变。另外还可显示脑发育不良、脑萎缩等病变。
治疗原则:BH4缺乏症的治疗主要取决于酶缺乏类型及脑脊液中神经递质缺乏程度。大多数BH4缺乏症都需要神经递质前质多巴及5-羟色氨酸联合治疗。PTPS轻型者可单纯BH4治疗,但需要密切随访神经系统症状;严重型者给予BH4联合神经递质前质治疗。DHPR缺乏症者目前认为BH4治疗可导致7,8-二氢生物蝶呤堆积,对芳香族氨基酸羟化酶及NO合成酶产生负面影响,建议采用饮食治疗降低血Phe浓度,同时需要神经递质前质及四氢叶酸治疗。GTPCH缺乏症伴有HPA者需要BH4联合神经递质前质治疗。
1.四氢生物蝶呤治疗
主要目的是降低血苯丙氨酸浓度。PTPS缺乏症、GTPCH缺乏症、PCD缺乏症患者在普食下给予BH4(10mg/片)1~5mg/kg,分2次,空腹服用;科望(盐酸沙丙蝶呤片、100mg/片)2~5mg/kg,每天1次,早餐进食时服用。根据血Phe浓度调节剂量,有些患者给予BH41~2mg/(kg·d)治疗,即可使血Phe浓度维持正常水平。
2.神经递质前质治疗
大多数BH4缺乏症都需要神经递质前质多巴(L-DOPA/Cabidopa)及5-羟色氨酸(5-HTP)联合治疗。临床上多用美多巴或息宁(L-DOPA/Cabidopa=4∶1)。左旋多巴、5-羟色氨酸宜从1mg/(kg·d)开始,每周递增1mg/(kg·d)剂量(表1)。
表1各年龄段患儿神经递质前质治疗剂量

引自:遗传代谢病防治理论与实践.第1版.ISBN:978-7-117-34392-3.主编:
为减少药物所致的胃肠道不良反应或药物不耐受,L-DOPD及5-HTP药物开始治疗剂量从1mg/(kg·d),每周增加1mg/(kg·d),至治疗剂量。如对多巴不良反应可出现运动障碍、兴奋失眠等,尤其是儿童患者初始治疗时易发生,减少多巴剂量或总量分多次服用可改善上述症状;此外,L-DOPA治疗中往往会出现On-Off现象,即间歇性出现精神萎靡不振、软弱无力、嗜睡等,可在1天中出现几次,这种精神运动状态改变与较短的L-DOPA半衰期有关,将1天药物总剂量分成6~8次服用可减少On-Off现象。5-HTP可导致呕吐、腹泻等肠胃道紊乱症状,重者可减少剂量或暂时性停药。
3.对于DHPR缺乏者,研究发现如给予大剂量BH4会导致7、8二氢生物蝶呤堆积,影响芳香化酶及一氧化氮合成酶作用。因此需用低或无Phe特殊奶粉或蛋白粉等饮食治疗,使血Phe浓度控制到接近正常水平(120~240µmol/L),方法同PKU。同时需要神经递质前体治疗:同PTPS型以及四氢叶酸(亚叶酸钙):10~20mg/d。
4.对有生育要求的BH4缺乏症高危家庭,可在先症者基因突变明确的基础上实施产前诊断,阻止患者出生。
PKU属常染色体隐性遗传,其特点是:①患儿父母都是致病基因突变携带者(杂合子);②患儿从父母各得到一个致病基因突变,是纯合子;③患儿母亲每次生育有1/4可能性为PKU患儿;④近亲结婚的家庭,后代发病率较一般人群为高。
1.避免近亲结婚。
2.对BH4D高危家庭产前诊断是优生优育,防止同一遗传病在家庭中重现的重要措施。对有本病家族史的夫妇及先证者可进行 DNA分析,并对其胎儿进行产前诊断。家族成员基因分析也可检出杂合子携带者,进行遗传咨询。
3.开展新生儿筛查,及早发现BH4D患儿,尽早开始治疗,减少并发症以及不良预后。
4.产前诊断
BH4D先证者的母亲若再次妊娠,可在妊娠16~20孕周时经羊水穿刺或10~12孕周经绒毛膜绒毛取样提取胎儿细胞的DNA,可对突变已知家系进行基因产前诊断。









