


 收藏
收藏 已收藏
已收藏英文名称 :inherited metabolic disorders
遗传代谢病(inherited metabolic disorders)是遗传病中的一组代表性疾病,是遗传性生化代谢缺陷的总称,迄今已命名900余种遗传代谢病。患者可自胎儿至老年发病,疾病导致单个脏器或多脏器损害,脑损害为常见或首发表现,症状及体征缺乏特异性,容易被延误诊断,致残率及致死率很高,需要高度重视。随着生化分析及医学遗传技术的进步,各类遗传代谢病的病因、发病机制、遗传方式逐步明确,筛查、诊断与治疗技术迅速发展,很多遗传病从不治之症成为可治可防的疾病,患者的生存质量显著提高。
遗传代谢病均为单基因遗传病,绝大多数为常染色体遗传病,以隐性遗传为主,少数疾病为常染色体显性、X连锁或线粒体基因遗传方式。由于疾病类型、缺陷程度、生活环境、诊断早晚的差异,患者临床表现各异,轻重不等,轻者可能终身不发病,重者在新生儿期死亡。临床医师需要提高警惕,重视症状、体征及一般检查中的线索,争取及早病因诊断,正确治疗。
遗传代谢病对机体的损害可表现为一个或多个方面:①代谢终末产物缺乏,机体所需产物合成不足或完全不能产生。如生物素酶缺乏导致肠道生物素吸收障碍、体内生物素运输障碍,多种羧化酶功能缺陷、有机酸血症。②前身代谢物蓄积,引起自身中毒、细胞及器官肿大、代谢紊乱。如甲基丙二酸血症患者体内蓄积的甲基丙二酸及其代谢物有严重的神经毒性,糖原贮积症Ⅰ型患者糖原分解受阻,引起肝大、低血糖和能量代谢障碍。③旁路代谢途径加强。如高苯丙氨酸血症,患者体内除了苯丙氨酸蓄积外,旁路代谢形成大量苯丙酮酸、苯乙酸,产生神经毒性。④生理活性物质合成障碍,如21羟化酶缺乏症导致皮质醇合成障碍、肾上腺皮质功能不全、肾上腺皮质增生症。⑤物质转运功能障碍。细胞膜有主动转运系统,如肠道黏膜铜吸收障碍,引起Menkes病;肾小管重吸收障碍,引起Hartnup病、肾小管酸中毒、磷酸尿性佝偻病等。
基因突变导致蛋白质结构缺陷或合成、分解代谢异常,引起相应的组织病理损害和临床症状。有缺陷的物质可能是一个复杂的大分子蛋白,也可能是一个较简单的小分子,或是一个细胞器。由于生化反应阻滞,相关物质在体内的合成、代谢、转运和储存等环节出现异常。
根据异常代谢物的分子大小,可将遗传代谢病分为两类,①小分子病:例如氨基酸代谢病、有机酸代谢异常、线粒体脂肪酸代谢病、单糖类代谢病、金属代谢异常等,一些患者起病较早,严重者可在出生后数分钟发病,表现为急性重症脑病或多脏器损害,甚至猝死;轻者可能终身不发病。②细胞器病:又称大分子病,代谢物沉积在细胞及组织中,如糖原贮积症、脂类代谢病、黏多糖贮积症、糖蛋白病、线粒体病、过氧化物酶病等,常在婴幼儿期或儿童期起病,病程多为慢性进行性变性过程。
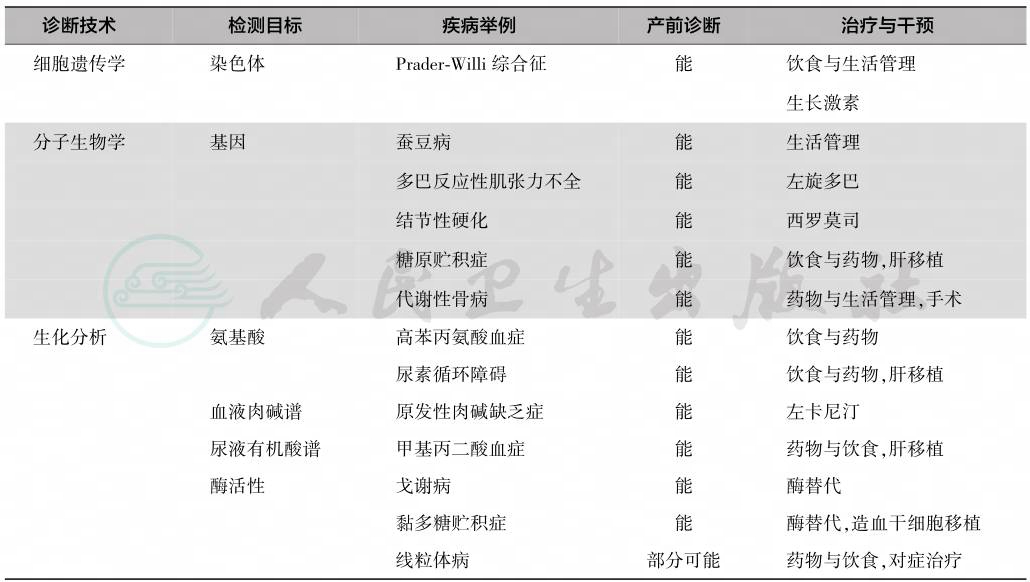
根据受累物质的特点,遗传代谢病可分为下列几种(表1)。
表1 遗传代谢病的主要类型和病种

虽然多数遗传代谢病缺乏有效的治疗方法,随着研究的进展,能治疗的疾病在逐步增加,通过对因和对症治疗,可以阻止病程,减轻症状。
遗传代谢病总的治疗原则为补其所缺、排其所余、禁其所忌,针对疾病可能造成或已经造成的器官损害进行干预,根据不同的病种和患儿个体情况选择相应的方法,通过饮食、药物、移植治疗进行干预(表2)。
表2 不同类型的遗传代谢病需要的诊断与治疗技术
(一)饮食治疗
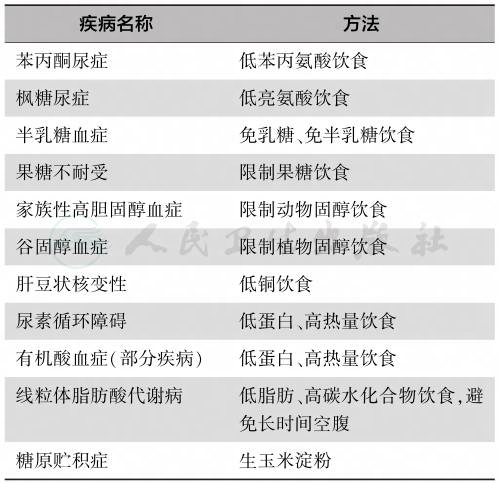
1953年德国Bickel医师创立了遗传代谢病的饮食疗法,通过低苯丙氨酸饮食治疗有效地降低了一位苯丙酮尿症(phenylketonuria,PKU)女童血液中的苯丙氨酸浓度,神经精神症状随之改善。此后,借鉴PKU的饮食治疗原理,国内外逐步建立了氨基酸、有机酸、脂肪酸、碳水化合物等多种代谢病的饮食治疗方法(表3)。
表3 遗传代谢病的饮食治疗方法
饮食治疗的原理为限制代谢障碍前驱物质摄入,减少毒性代谢物产生,同时要保证生长发育及生理活动所需要的热量、蛋白质、脂肪、维生素、矿物质等各种营养素。即使是相同疾病的患儿,由于脏器损害及酶缺陷程度的不同,对于各种食物的耐受能力及营养素的需求也不同,个体化饮食指导至关重要。
(二)药物治疗
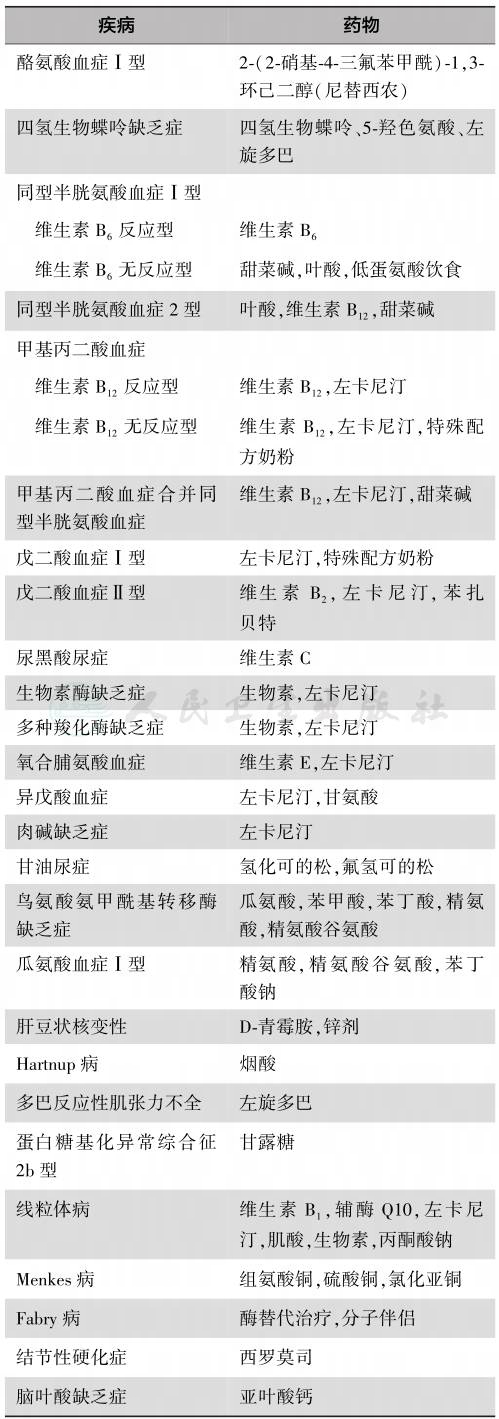
对于部分遗传病,可采用维生素、辅酶、激素等药物进行治疗,促进有害蓄积物的排泄,补充生理活性物质(表4)。对于特纳综合征、Prader-Willi综合征、Noonan综合征,自婴儿期给予患儿生长激素支持,特纳综合征患儿于青春前期开始雌孕激素补充治疗,绝大多数患儿可以获得良好的体格及智力发育。
近十余年来,气相色谱质谱法尿液有机酸分析、液相串联质谱法血液氨基酸及酰基肉碱谱分析已经成为我国遗传代谢病筛查与诊断的主要技术,氨基酸、有机酸、脂肪酸代谢病的治疗经验逐步成熟,通过药物与饮食治疗,避免脑、肝、心脏等重要脏器损害,患儿生存质量显著改善。随着高危筛查的普及,临床医师对于遗传代谢病的识别能力逐步提高,越来越多的患儿被发现,一些患儿获得了正确诊断与治疗。甲基丙二酸血症、丙酸血症、生物素酶缺乏症等有机酸代谢病受到了儿科、围产医学、神经科、精神科领域的高度重视,通过临床高危筛查,很多新生儿期到成人期的患者获得了正确诊断与治疗,预后良好。在先证者生化与基因诊断明确的基础上,很多遗传病可以进行产前诊断,帮助相关家庭避免疾病再发的风险,生育健康后代。
表4 遗传病的药物治疗
线粒体病的治疗也取得了进步。经典线粒体病以鸡尾酒疗法(维生素B1、辅酶Q10、左卡尼汀、中链脂肪酸等营养素支持)为主,精氨酸、丙酮酸钠、肌酸对线粒体基因3243位点突变患儿治疗有效,丙酮酸脱氢酶复合物E1α亚单位缺陷患儿大剂量维生素B1疗效良好,生物素及维生素B1反应性脑病患儿经生物素及维生素B1治疗后显著好转。
溶酶体存在于人体各种细胞的胞质内,是细胞的消化器官,含有50多种酸性水解酶,分别参与糖蛋白、脂蛋白、多糖、黏多糖、黏脂、核酸等基质的分解代谢。黏多糖贮积症Ⅰ型、戈谢病、糖原贮积症Ⅱ型、Fabry病等少数溶酶体病酶替代治疗方法成熟,一些疾病可以通过小分子伴侣药物改善病情。对于异染性脑白质营养不良、尼曼-皮克病,一些国家在进行酶替代治疗研究,在神经系统严重受损之前,可以争取造血干细胞移植。
在其他遗传代谢病的药物治疗方面,国内外亦获得了可喜的研究进展。如西罗莫司在结节性硬化症的治疗中疗效显著。甘露糖对蛋白糖基化异常综合征2b型疗效良好。
1.祛除有害物质
针对高氨血症,苯丁酸钠、苯甲酸钠可促进氨的排泄。针对酪氨酸血症,早期口服2-(2-硝基-4-三氟苯甲酰)-1,3-环己二醇(尼替西农)治疗效果良好。左卡尼汀是线粒体脂肪酸β氧化循环的关键维生素,是有机酸、脂肪酸代谢病治疗的基本药物,不仅有助于纠正急性酸中毒,也可有效地改善远期预后。D-青霉胺可与铜结合,促进铜的排泄,对多数肝豆状核变性患儿有效。硫酸锌、醋酸锌等锌剂可阻止肠道铜的吸收,减少铜的蓄积,可减少D-青霉胺剂量,提高肝豆状核变性的治疗效果。
2.维生素疗法
很多维生素作为辅酶参与物质代谢,除了先天性酶缺陷以外,一些疾病为辅酶代谢障碍所致。某些维生素对于遗传代谢病患儿为关键治疗药物,如生物素对于生物素酶缺乏症和全羧化酶合成酶缺乏症患儿有戏剧般治疗效果,维生素B12对维生素B12反应型甲基丙二酸尿症、维生素B2对于部分戊二酸血症Ⅱ型疗效良好(表4)。
3.补充缺乏的生理活性物质
由于吸收障碍、生成不足、消耗增多,遗传代谢病患儿体内常缺乏一些生理活性物质。如四氢生物蝶呤合成酶缺乏症患儿低苯丙氨酸饮食无效,需要长期补充四氢生物蝶呤、5-羟色氨酸、左旋多巴等神经递质前质。Menkes病患儿肠道铜吸收障碍,体内铜缺乏,需要皮下注射组氨酸铜、硫酸铜或氯化亚铜。鸟氨酸氨甲酰基转移酶缺乏症和氨甲酰磷酸合成酶缺乏症患儿需要长期补充瓜氨酸,而瓜氨酸血症患儿则需要补充精氨酸。
(三)酶替代治疗
1.血浆、血球
细胞生物化学研究证实,溶酶体酶可从淋巴细胞转移到成纤维细胞中,黏多糖贮积症、神经鞘脂病、糖原贮积症Ⅱ型可通过输注健康人血浆暂时改善患儿病情。静脉滴注红细胞悬液可改善重症精氨酸酶缺乏症患儿尿素循环,缓解症状。
2.酶替代治疗
近年来,分子生物学技术应用于酶的纯化生产,酶替代治疗在溶酶体病的治疗中取得了巨大成功。如戈谢病Ⅰ型、Fabry病、黏多糖贮积症Ⅰ型、糖原贮积症Ⅱ型,通过定期静脉注射补充所需的酶,可有效控制疾病进展。
(四)细胞或器官移植
同种器官移植可以提高患儿体内酶的活性,并导入正常的遗传信息,有时可以修正患儿器官功能。骨髓造血干细胞移植在遗传代谢病的治疗中应用最为广泛,如黏多糖贮积症、过氧化物酶体病、地中海贫血、腺苷脱氨酶缺乏症等疾病,预后良好,早期骨髓移植是挽救生命的关键方法。全肝移植或活体部分肝移植是治疗尿素循环障碍、糖原贮积症Ⅰ型、家族性高胆固醇血症、肝豆状核变性、酪氨酸血症等疾病的重要手段,国内外取得了成功的经验。近年来,干细胞技术也开始应用于一些遗传病的治疗,有望使更多的患儿受益。
(五)基因治疗
从理论上讲,基因治疗是治疗各种单基因遗传病最理想的方法,腺苷脱氨酶缺乏症、镰状红细胞病、脊髓性肌萎缩症、进行性肌营养不良等疾病的基因治疗取得了成功。但是,基因治疗受多种物理、化学、伦理因素的影响,面临很多困难,与骨髓移植相比难度更大。
(六)急性期治疗
部分氨基酸、有机酸、脂肪酸代谢病患儿以急性形式起病,合并酮症、代谢性酸中毒、低血糖、高氨血症等严重代谢紊乱,多脏器损害,严重时猝死。根据不同的病种应给予静脉补液、药物与饮食治疗、对症治疗,必要时需血液净化治疗。一些患儿既往无异常病史,因“发热、腹泻、呕吐、饥饿、疲劳、暴饮暴食、预防接种”等应激因素或药物诱发急性发作,导致瑞氏综合征、心肌病,甚至猝死,易引发医疗纠纷。如果患儿在病因不明的情况下死亡,无法对家族成员进行正确的遗传咨询与健康指导,同胞可能再次发生类似灾难。留取尿液、血液或细胞样本,进行生化代谢与基因分析,有助于进一步病因分析。
(七)治疗时期
1.出生前治疗
如母性PKU,随着新生儿筛查的普及和治疗方法的成熟,各国已经有很多PKU患儿长大成人,结婚生育。经过治疗发育正常并同健康男性结婚的PKU女患者,如果孕前、孕期不合理地控制饮食,胎儿流产、死产、畸形、宫内发育不全等发生率很高,出生后多有小头畸形、智力低下、癫痫、先天性心脏病等合并症。因此,对育龄期女性PKU患者应进行饮食管理,最好在孕前半年开始治疗直至分娩,使血苯丙氨酸浓度控制在 120~360μmol/L,以保护胎儿。
2.症前治疗
很多疾病一旦发病,将造成难以逆转的脑损害或其他脏器功能损害,治疗越早,疗效越好。因此,对于少数治疗方法简单、筛查技术成熟的疾病,应进行新生儿筛查,争取在症状前确诊并开始治疗,以避免重要脏器损害,保证患儿健康成长。如苯丙酮尿症、原发性肉碱缺乏症、中链酰基辅酶A脱氢酶缺乏症,通过新生儿筛查或高危筛查帮助患儿获得早期诊断,在发病前开始治疗,则可预防脑损害、肝损害、心肌损害。
(八)其他治疗
很多遗传病患儿出生时或确诊时已经存在不同程度的脏器损害或肢体残障,需要综合干预,对症治疗,康复训练,必要时手术矫形。如对于肝损害的患儿给予保肝药物,高脂血症患儿需要降脂治疗,智力障碍或运动障碍的患儿需要语言、运动及认知训练。先天性骨病、肌营养不良患儿容易出现骨折、肢体畸形、脊柱畸形,需要给予支具防护,延缓功能性损害。









