


 收藏
收藏 已收藏
已收藏英文名称 :fragile X syndrome
一种X连锁智力障碍疾病。是引起智力障碍和孤独症谱系障碍最常见的单基因病,发病率仅次于21三体综合征,男性人群中约1/4 000,女性中约1/6 000~1/8 000。因患者的体细胞在低叶酸培养基中,经诱变剂作用特殊制备染色体,可在部分细胞的染色体Xq27.3位点出现如同断裂的脆性部位而得名。是第一个被先发现的TREDs。
FXS的发病原因是脆性X智力障碍基因(fragile X mental retardation gene 1,FMR1)的 5′非翻译区遗传不稳定的CGG三核苷酸重复序列异常扩增以及相邻CpG岛的异常甲基化。
FMR1定位于染色体Xq27.3,长38kb,含17个外显子;有多种剪接方式,产生不同的mRNA。FMR1第1外显子的5′非翻译区有一段CGG三核苷酸重复序列,在其上游约250bp左右有CpG岛。正常人群CGG重复数存在多态性,重复的次数6~53,一般为30左右,CGG重复之间常含2~3个拷贝的AGG重复,称为AGG中断,这种重复序列的结构在亲代向子代传递过程中保持稳定。但当CGG重复的次数超过55或AGG中断丢失成为连续CGG重复扩增大于35~40次,成为不稳定重复序列,被称为前突变(premutation)。在正常人群中有1%的人CGG重复40~54次或CGG重复35~45次而无AGG中断,这种序列有不稳定的趋势,被称为灰色区(grey zone),稳定程度很难预测;CGG重复次数大于200为全突变(full mutation)。全突变的男性患FXS,女性由于存在X染色体失活的影响表型复杂。重复次数与表型关系见表1。
表1 脆性X综合征CGG重复扩增和相关表型
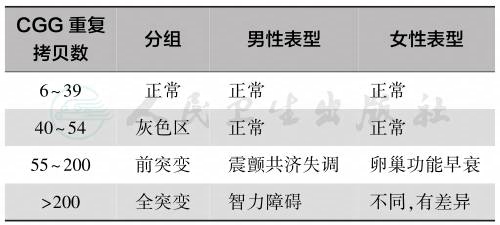
当CGG重复次数大于55,基因变得不稳定,在亲代向子代传递过程中CGG重复数可能发生变化:携带前突变的母亲传递到子代后可使子代成为全突变;母亲是全突变携带者可传给子代一个更大或缩小的全突变;携带灰色区等位基因的母亲可能传递给子代后使子代成为前突变,但不会成为全突变。相反,父亲无论是前突变携带者或是极罕见的全突变(全突变的男性罕有生育)只能将前突变传递给子代。
几乎所有FXS由三核苷酸重复突变所致。少见有FMR1缺失突变,缺失大小1bp至数百或百万个碱基;点突变或重复等均有报道。
脆性X智力障碍蛋白(fragile X retardation protein,FMRP)是FMR1的翻译产物,它是一种胞质RNA结合蛋白,含RNA结合区、细胞核输出信号和细胞核定位信号,功能是与特定的mRNA结合将其从核运输到胞质,功能与调节神经发育的特异基因表达和神经元突触可塑性有关。FMR1在全突变时扩增的CGG重复序列和CpG岛的高度甲基化抑制FMR1转录,使其不能产生FMRP,FMRP的缺失引起FXS。女性因仍有一条正常X染色体,其FMR1可产生FMRP,故无或仅有轻度异常。FMR1在前突变状态下通常CpG岛无高度甲基化,FMR1转录活跃,产生比正常人高的mRNA,但翻译效率低,所以FMRP产量低。此外,还存在突变的嵌合体:体细胞有前突变和全突变两个细胞系,即CGG重复长度嵌合;有高度甲基化和非甲基化的FMR1,即CpG岛甲基化嵌合。这些也可能与智力障碍的程度有关。
邻近FRAXA还有FRAXE及FRAXF两个脆性部位,均有CGG重复序列。FRAXE的基因为FM R2,可因CGG重复扩增抑制表达而出现表型异常,命名为脆性XE综合征(fragile XE syndrome),临床上 FRAXE与FRAXA相似,但较轻,目前尚无在中国人群中发现本型疾病的报道。FRAXF无病理意义,与家族性的智力障碍无关。
1.细胞遗传学脆性部位检查
因患者只有4%~40%的细胞能显示脆性部位,而且有假阳性及假阴性出现,目前已基本不使用。
2.Southern印迹杂交
提取外周血基因组DNA,用不同的限制性内切酶,包括甲基化敏感的内切酶消化DNA(常用EcoR1和Eag1),经琼脂糖凝胶电泳、转膜和与标记的DNA探针(StB12.3)杂交后分析CGG重复的片段长度和甲基化状态。该方法目前仍然是国际公认的金标准。能够鉴别前突变和全突变的等位基因并提供甲基化的信息。
3.PCR扩增
特异性扩增FMR1的CGG重复序列,经毛细管电泳准确检测DNA片段的长度,计算出CGG的重复次数。可以检出前突变、全突变及嵌合状态;还能判断女性基因的杂合或纯合性以及AGG中断。
无有效方法,语言训练对患儿相当助益。其他科学的训练有助于患儿掌握生活技能,提高注意力等。
FSX是X连锁不完全显性遗传病,患儿的母亲是前突变或全突变携带者。









