


 收藏
收藏 已收藏
已收藏人类疾病由内因与外因即先天遗传因素与环境因素单独或相互作用所致。在小儿时期,遗传及先天因素尤居首要。2013年9月国家卫生计生委公布我国每年有1600万婴儿出生,约有90万带有出生缺陷,中国出生缺陷总发生率约为5.6%,每年临床上明显可见的出生缺陷约25万例,围产期出生缺陷总发生率呈上升趋势。出生缺陷(birth defects)或先天异常(congenital anomalies)是指胚胎或胎儿期在发育过程中所发生的形态结构或功能异常,遗传病是其中重要的组成部分。遗传病主要分为:①染色体病:由整条或部分片段缺失、重复或其他改变所致,如21-三体综合征、Turner综合征和猫叫综合征;②单基因病:由一个等位基因突变所致,孟德尔遗传病主要指这类疾病,如苯丙酮尿症、结节硬化症、Duchenne肌营养不良和肌强直性营养不良;③多基因病:多个基因与环境共同作用,如出生缺陷中的唇腭裂和一些成年人的心脑血管病和糖尿病;④线粒体病:由线粒体DNA突变所致;⑤体细胞遗传病:肿瘤。
各类遗传病在不同群体中发病情况不同。在孕早期自发性流产的胚胎中,50%由于染色体异常,以染色体数目异常为主。人群普查中显示:染色体病有300余种,约占0.5%~1.0%;单基因病近6000余种,每种发病率均不高,但由于种类多,总的发病率并不低,约占2%(表1)。多基因病也称复杂疾病,与遗传和环境均有关,包括一些先天异常和常见病,发病率较高,可达15%~20%。各类遗传病在人群中总的发病率约20%~25%,占总人口的1/5~1/4,在我国,估计达到2.5亿~3亿人之多。一些成年期后始发的遗传病在婴幼儿时即已初露端倪;许多遗传病为小儿期或围生期死亡的首要原因,占总死亡率40%以上;即使后天性疾病如传染病也有易感性,外伤后也有愈合快慢、出血多少的问题,这些都受遗传因素影响;尽管表型正常的人,也并非无遗传方面的问题,每个个体都携带有6个左右隐性的有害基因(或称遗传负荷),本人虽不患病,却可向后代传递,成为致病基因携带者。现代社会又增加了工农业、城乡环境、空气、水源的污染,导致新生性突变发生,使遗传病和先天畸形成为儿科领域中一个突出的问题。阐明遗传病的遗传病理机制,研究防治方法,早防早治、保障儿童健康,儿科医生的责任殊为重要。
表1 人类孟德尔遗传数据库统计资料*
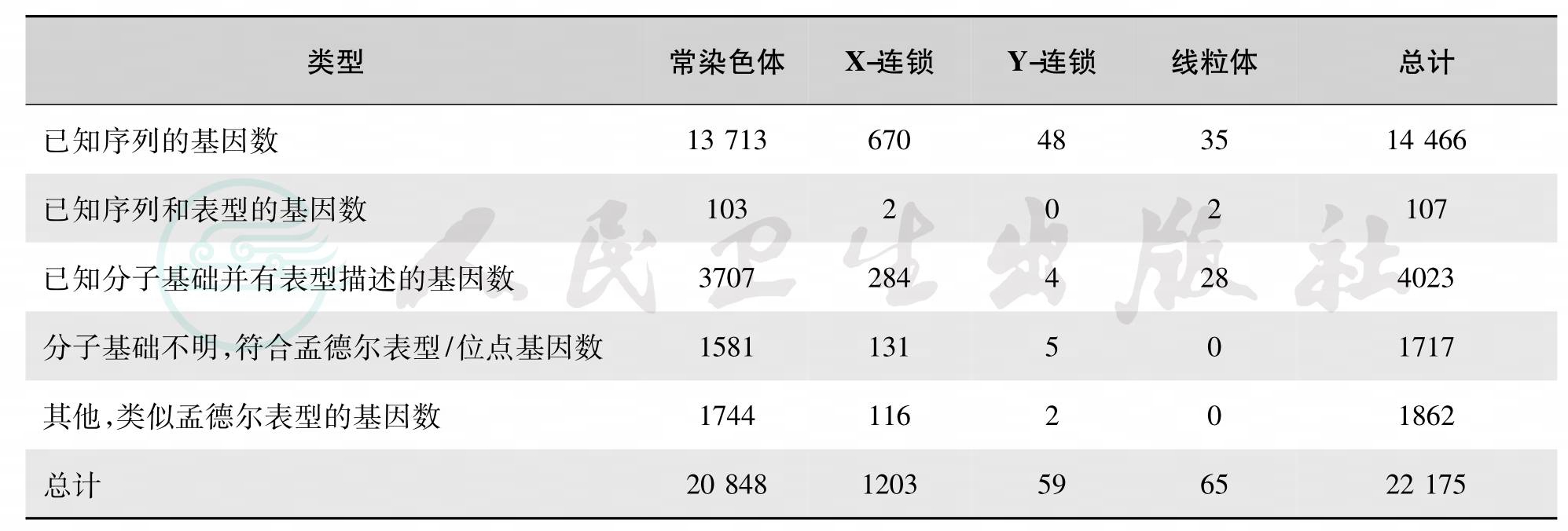
注:*2014年1月22日
一、遗传的基本概念
各种生物都能通过生殖产生子代,子代和亲代之间,不论在形态结构和生理功能的特点上都很相似,这种现象称为遗传(heredity)。但是亲代和子代之间,子代各个体之间不会完全相同,总会有所差异,这种现象称为变异(variation)。研究生物遗传变异规律的一门科学,称为遗传学(genetics)。医学遗传(medical genetics)则是将遗传学的基础理论与临床医学相结合的一门新兴科学,随着医学遗传学的发展,产生许多分支,如细胞遗传学(cytogenetics)、分子遗传学(molecular genetics)、免疫遗传学(immunogenetics)、肿瘤遗传学(tumor genetics)、群体遗传学(popular genetics)、行为遗传学(behaviour genetics)、药物遗传学(drug genetics)、发育遗传学(developmental genetics),以及与各器官相关的遗传病学等,均与儿科密切相关。
遗传学以研究染色体和基因为起点,染色体和基因是遗传的物质基础。
(一)染色体是遗传信息的载体
1.染色体的数目和形态
染色体(chromosome)位于细胞核内,数目和形态稳定。人类体细胞的染色体数目为23对(46条),每一对染色体中的一条来自父亲,另一条来自母亲。23对中22对男性与女性相同,称为常染色体(autosome),还有一对决定性别,称为性染色体(sex chromosome):女性为XX,男性为XY。正常男性的Y染色体遗传自父亲,X染色体遗传自母亲;正常女性的两条X染色体分别遗传自父亲和母亲。这种在生物学上具有成对染色体的细胞,称为二倍体(diploidy),用2N表示。成对的常染色体称为同源染色体(homologous chromosome),携带的基因非常相似;X和Y染色体不是同源染色体。染色体在细胞周期中以不同的形态存在,有丝分裂中期轮廓结构清楚,形成光学显微镜可以看到的染色体,如图1。
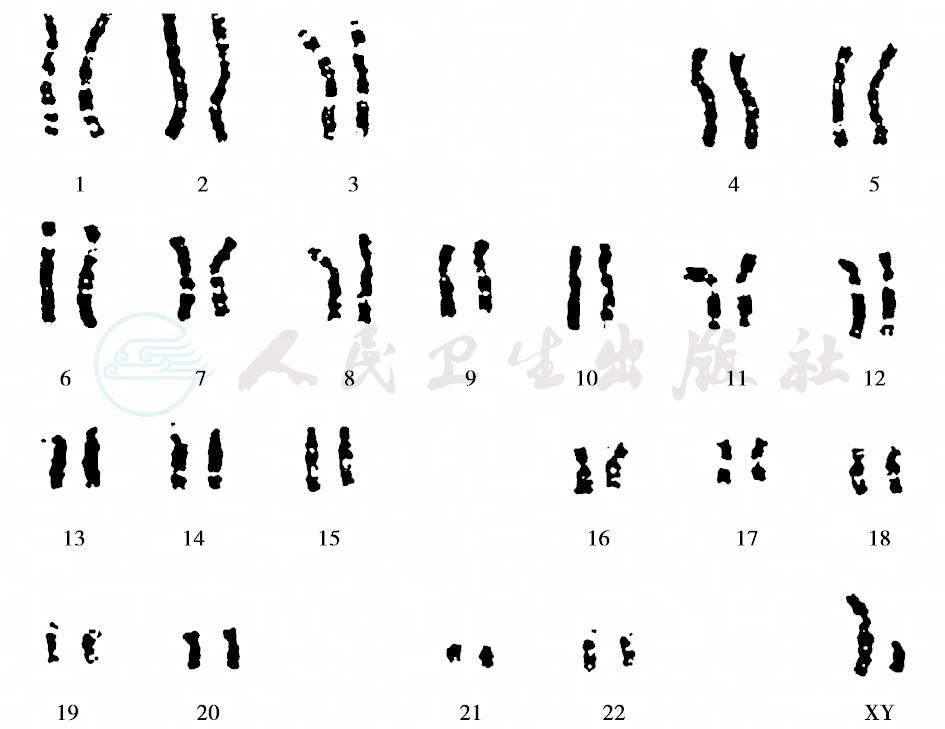
图1 中国人正常染色体组型及带型
图中的染色体是在细胞分裂的中期所看到的,所以每个染色体已经形成两条染色体,在染色体两臂之间就是着丝点所在处。如果着丝点不在染色体的中央,而偏于一端,则使染色体分成长短两份。如着丝点位于染色体的近末端,这种染色体称为近端着丝点染色体。人类的第13~15、21、22染色体共五对,是近端着丝点染色体,在其前端都有一个小体,称为卫星或随体(satellite)
一条完整的染色体包括着丝粒(centromere)、染色体臂和端粒(telomere)。着丝粒是染色体经染色后不着色或浅着色的狭窄部位,也称为主缢痕(primary constriction),它把染色体臂分成短臂(short arm,p)和长臂(long arm,q);位于末端的端粒能够保持染色体构型稳定而不相互融合。短臂和长臂分为若干区,区中有带(如2q23,即指2号染色体长臂2区3带)。各区带中含有众多基因(gene),如图2。
着丝粒位置恒定,染色体按其位置分为中着丝粒染色体:着丝粒位于染色体中央,短臂和长臂大致相等;亚中着丝粒染色体:着丝粒近于染色体一端,短臂短于长臂;近端着丝粒染色体:着丝粒靠近染色体末端,短臂很短,长臂长,短臂末端有圆形或略呈长形的突出体被称为随体(satellite),随体由随体柄将其与短臂连接,随体柄与核仁形成有关,称核仁组织区(nucleolus organizer region,NOR)。
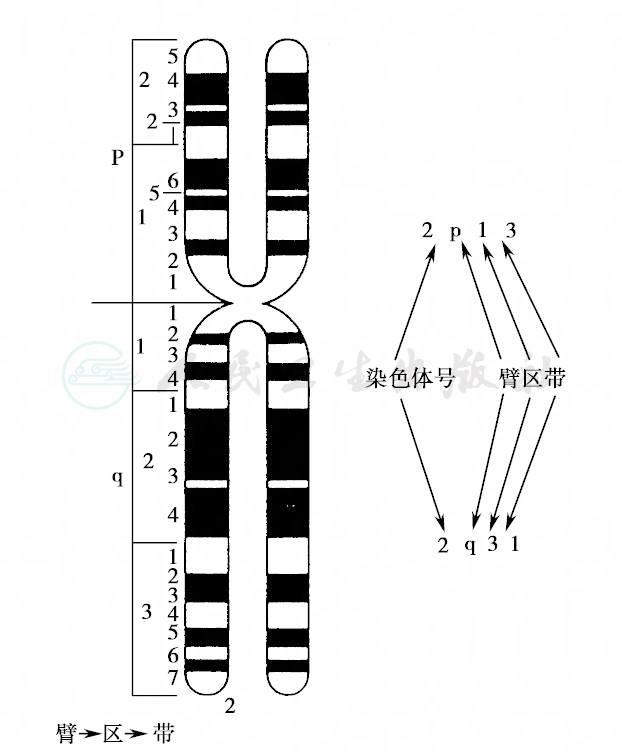
图2 显带染色体的界标、区和带示意图
空白部分:Q带的暗带,G带的浅染带
黑色部位:Q带的亮带,G带的深染带
2.染色体的化学组成和结构单位
染色质和染色体是同一种物质的不同存在形式,在间期细胞核内被称为染色质,当细胞进行分裂时染色质高度螺旋化、紧密盘绕和折叠成为染色体。
染色质分为常染色质(euchromatin)和异染色质(heterochromatin)。G显带时,常染色质浅染,含有具转录活性的基因,是基因转录活跃部位;异染色质深染,又分为结构性异染色质(constitutive heterochromatin)和兼性异染色质(facultative heterochromatin)。结构性异染色质主要分布于着丝粒及周围、端粒和某些染色体的区段,所含的DNA主要是一些简单的非高度保守的高度重复序列,没有转录活性;兼性异染色质在一定条件下可以转变为常染色质恢复转录功能,如女性两条X染色体中失活的一条X染色体等。
每一条染色体由一个线性的、完整的双螺旋脱氧核糖核酸(deoxyribonucleic acid,DNA)分子和围绕其中的组蛋白(histone)和非组蛋白(non-histone)组成。组蛋白是一类碱性蛋白质,含精氨酸和赖氨酸等带正电荷的碱性氨基酸较多,分为H1、H2a、H2b、H3、H4五种亚型,与DNA双股螺旋分子中带负电荷的磷酸基团相互结合形成染色质的初级结构。组蛋白作为DNA构型的核心颗粒,变化甚少。非组蛋白是染色质内组蛋白以外的所有蛋白质,含天门冬氨酸和谷氨酸等酸性氨基酸较多,包括DNA和RNA聚合酶、转录酶、解旋酶、逆转录酶、端粒酶,以及参与基因调节的有关因子、作用于组蛋白的一些酶,如组蛋白甲基化酶等,以及影响染色体和染色质构象的蛋白和其他有关的酶。
染色质的基本结构单位是核小体(nucleosome)(图3),由核心颗粒和连接丝组成。核心颗粒由各两分子的4种组蛋白H2A、H2B、H3和H4组成的8聚体及在外围盘绕13/4圈的链长140~150个核苷酸的核心DNA结合形成的球形结构;连接丝主要包括组蛋白H1、长度为20~60个核苷酸的DNA链和一些非组蛋白。核小体通过DNA分子连接形成串珠结构,构成染色体的一级结构;一级结构进一步螺旋化,每6个核小体以组蛋白H1为中心形成一个螺线管(solenoid),构成染色体的二级结构;120个螺线管进一步螺旋化,形成染色质环(chromatin loops),构成染色体的三级结构,大约100kb;染色质环进一步盘绕、折叠构成染色单体;经过高度压缩人类细胞中约2米长DNA链容纳于直径6μm的细胞核中。
3.细胞周期、有丝分裂和减数分裂
细胞周期是指细胞从前一次分裂结束开始到下一次分裂结束为止这样一个周期,分为两个阶段:间期(interphase)和有丝分裂期。一个细胞经过有丝分裂(mitosis)形成两个子细胞的过程称为细胞增殖。细胞周期中每条染色体正确地自我复制,形成两条姐妹染色单体,然后平均地分到新形成的两个子细胞中。所以,子细胞和母细胞的染色体数目相等。通过有丝分裂保证了细胞增殖过程中始终保持染色体数目和形态恒定,这是遗传性具有相对稳定性的细胞学基础。在形成配子(精子和卵子)时,通过减数分裂(meiosis),即染色体复制一次,细胞分裂两次,使成对的染色体减半,产生有单倍染色体的配子(单倍体,1N)。受精后,精子与卵子结合为合子,细胞内的染色体又重新组合成为二倍体(2N)。这样,个体的染色体能够在世代相传中保持稳定的数目,并且借此保持来自双亲的遗传特性。人类和大部分高等动物的生活史可简化如图4。
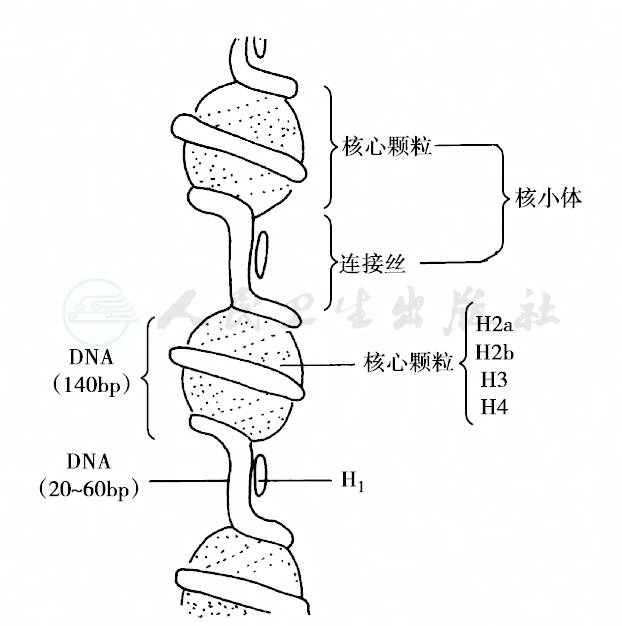
图3 核小体——染色质的基本结构单位
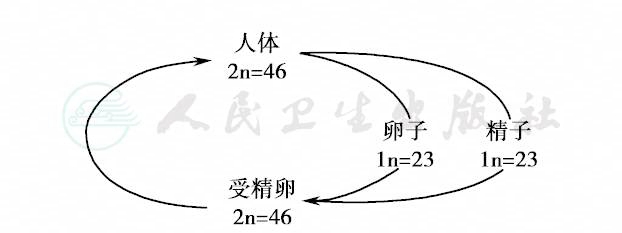
图4 人类和部分高等动物染色体的组合
4.染色体的制备和染色体核型
细胞核中的染色体在细胞有丝分裂中期形态最典型,原则上能够发生有丝分裂的各种组织细胞和悬液均可制备染色体,常用骨髓细胞、外周血淋巴细胞、皮肤成纤维细胞以及孕后的绒毛、羊水和脐带血细胞等进行体外培养。对不分裂的细胞加入植物血凝素刺激分裂,在培养后期加入少量秋水仙素,抑制细胞分裂时纺锤体的形成,使细胞分裂停止在中期,再用低渗溶液处理使细胞膨胀,染色体均匀分散,经过含乙醇和冰醋酸的固定液处理后将细胞悬液滴在玻片上进行干燥。显带技术常用胰酶消化后吉姆萨(Giemsa)染色显带,被称为G带,可显出400条带纹,用芥子阿的平(quinacrinemustard,QM)荧光染料所显示的带称为Q带。光学显微镜下分析染色体。
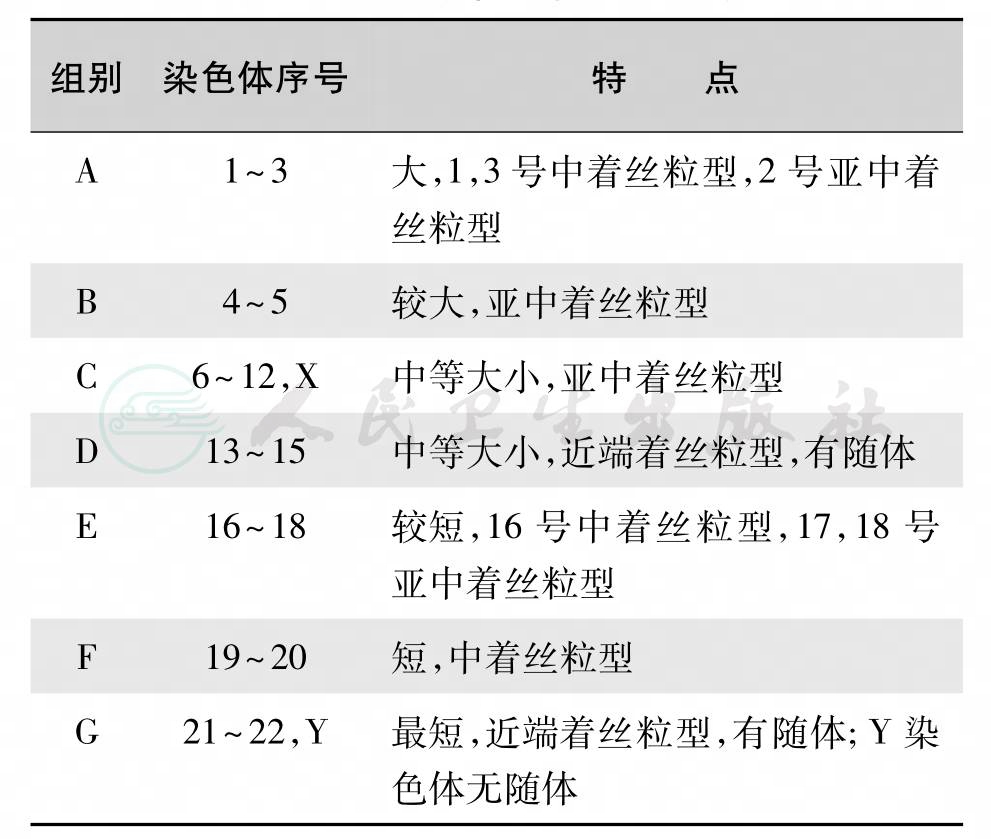
把一个体细胞的全部染色体按大小和形态特征有序地配对排列称为核型(karyotype)(图4)。根据核型对染色体的数目和结构进行分析的方法称为核型分析(karyotype analysis)。在显微镜下染色体长度不等,最长的1号染色体大约是最短的21号的5倍大。根据形态、相对长度、臂的比率、着丝粒的位置和随体的有无把23对染色体顺序地排列起来,常染色体从1到22编号并分为A、B、C、D、E、F、G 7组。性染色体X,属于C组,Y染色体归于G组(表2)。正常人体染色体核型的命名按照人类细胞遗传学国际命名委员会(International Standing Committee on Human Cytogenetic Nomenclature,ISCN)所规定的人类细胞遗传学国际命名体制(an international system for human cytogenetic nomenclature,ISHCN)进行。完整的染色体核型书写包括三个部分:染色体的总数、性染色体组成和染色体异常。如正常女性核型46,XX;正常男性核型46,XY。
表2 人的染色体分组和特点
染色体的数目和形态相对稳定是负载着遗传信息相对稳定的基础。如果一个个体的染色体数目增加或减少、一条染色体的片段增多或缺失都可能导致先天异常,如多了一条21号染色体引起Down综合征;5号染色体短臂末端缺失引起猫叫综合征等。染色体数目或结构畸变所引起的疾病称为染色体病。
5.染色体分析方法
染色体核型分析是细胞遗传学最基本的方法,细胞分裂中期或早中期,G显带显示400~550条带,DNA的分辨率5~10Mb。随分子遗传学技术的发展,以原位杂交为基础的细胞分子遗传学近年发展迅速,分辨率越来越高。常用的方法有:①荧光原位杂交(fluorescence in situ hybridization,FISH):用荧光标记特异的基因或染色体片段作为探针与滴在玻璃片上的染色体或间期细胞核进行杂交。中期染色体FISH分辨率为几个Mb,用前中期染色体分辨率可达到1Mb,用人工拉长DNA纤维FISH(DNA Fiber FISH)分辨率可达到几个kb。这些方法可用于诊断微缺失综合征、基因定位或帮助确定复杂的染色体重排;用非分裂细胞能够快速诊断染色体数目异常综合征。②多色FISH(M-FISH)和光谱染色体核型(spectral karyotyping,SKY)以24种不同颜色表现出22条常染色体和2条性染色体,在一次实验中观察到每一条染色体,具有更高的分辨率和敏感性,特别是在确定复杂核型和鉴定未知标记染色体或衍生的染色体来源有重要作用。③比较基因组杂交(comparative genomic hybridization,CGH)和微阵列CGH(array-CGH)用于分析人类基因组结构、基因定位和检出DNA拷贝数异常,分辨率可达到kb。④多重连接依赖的探针扩增(multiplex ligation-dependent probe amplification,MLPA)基于PCR反应,检测基因组DNA的拷贝数异常,一个反应中可以同时检测40个以上的靶点,方法简单、高效,比较适合于一般的临床诊断和大样本的筛查,主要用于染色体数目异常、亚端粒重排、诊断微缺失、重复综合征以及不明原因的原发性智力低下病因学检测。
(二)基因是遗传的物质基础
基因呈线型排列在染色体上,是实现遗传功能的基本单位,具有制约和决定人体性状的遗传信息。染色体是成对的,因此,基因也是成对地位于相对应的染色体上,这种成双相对的基因称为等位基因(allele)。
1.基因的分子结构
基因以DNA的化学形式存在于染色体上,作为遗传物质储存大量遗传信息。精确编码细胞生长、分裂、分化和对内外环境反应的所有指令。
DNA分子由两条互相缠绕、方向相反的大分子多聚核苷酸组成,形成双股螺旋状的结构。螺旋外侧为多核苷酸链,由脱氧核糖和磷酸基通过酯键交替连接而成。螺旋内侧为碱基,其垂直于螺旋轴通过糖苷键与多核苷酸链的糖基相连。4种碱基有:嘌呤:腺嘌呤(A,adenine)和鸟嘌呤(G,guanine);嘧啶:胸腺嘧啶(T,thymidine)和胞嘧啶(C,cytosine)。两条多核苷酸链中嘌呤和嘧啶间借氢键连接,使DNA双链彼此互补,即A和T(两个氢键),C和G(三个氢键)组成碱基互补对,称为碱基对(base pair,bp)。DNA分子的两条链借助互补碱基对之间的氢键并联在一起。如一条链的碱基序列为5'-GACATTG-3',它的互补链的序列5'-CTGTAAC-3'(图5、图6)。
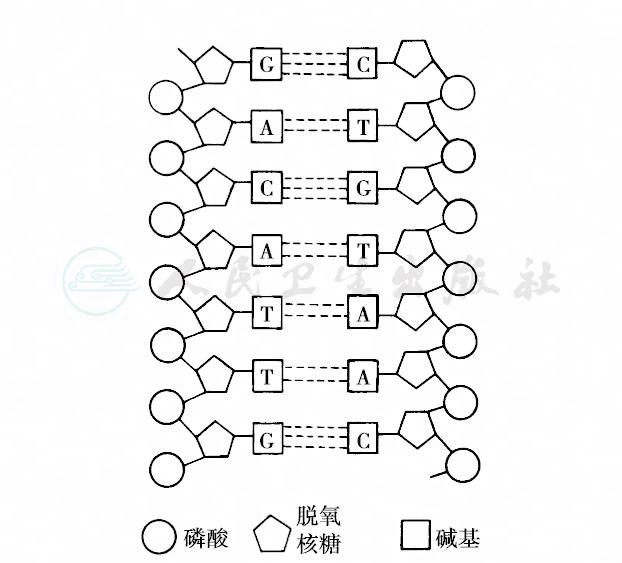
图5 DNA的结构
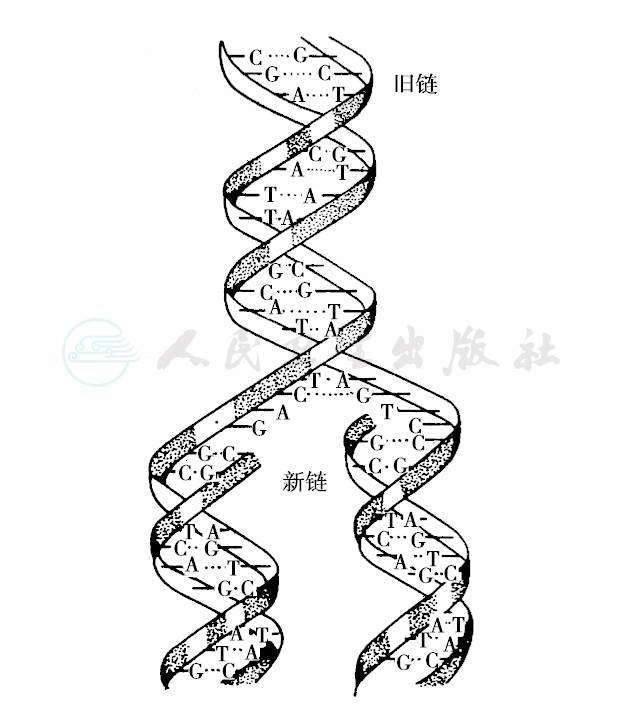
图6 DNA的“半保留”复制示意图
一个体细胞染色体所含DNA构成2个基因组,每个基因组的DNA含3.2×109 bp。基因组中DNA序列决定功能。随人类基因组计划研究的深入和结构基因组学的基本完成,推测人类基因组约有2万~2.5万个基因。这些与蛋白质合成有关的序列只占整个基因组序列的1.1%,剩余的绝大多数序列或为与基因及基因有关的序列,包括以单一序列或中等重复序列形式存在的位于非编码区的假基因、基因片段(如RNA基因序列和调控序列等)以及内含子和非编码序列;或为基因外DNA,包括单一或低拷贝或中等至高度重复DNA序列。
单拷贝DNA序列(single copy DNA)是人基因组中最重要的一类DNA,顾名思义,其在整个基因组中仅出现一次(或可能少数几次),大约占全基因组的45%。编码蛋白质的基因为单拷贝DNA,但它们仅占单拷贝DNA的一小部分,大部分单拷贝DNA是内含子或基因间的序列。基因组中其余的55%的序列为重复序列,可以数千次的重复,主要分两类:卫星DNA(satellite DNA)和散在重复序列(interspersed repeated sequence)。卫星DNA串联在一起,常位于染色体的特定部位;散在重复序列则散在全基因组。卫星DNA占全基因组的10%,可进一步分为三个亚类:①α卫星DNA,重复单位长度171bp,总序列长,分散在100kb至数个Mb之间的重复单位,聚集在染色体着丝粒异染色质区,一般不转录。重复单位常含特异着丝粒蛋白结合位点。②小卫星DNA(minisatellite),又称可变串联重复序列(variable number of tandem repeat,VNTR),重复单位6~64bp,长度0.1~20kb,多分布在端粒及其附近,绝大多数不转录。端粒DNA长3~20kb是串联的TTAGGG 6核苷酸重复,维持端粒的功能。③微卫星DNA(microsatellite DNA),重复单位2~6bp,又称短串联重复(short tandem repeat,STR),数量多,分散在基因组中。构成着丝粒、端粒、Y染色体长臂。二核苷酸重复最常见,如(CA)n、(GT)n、(AA)n、(GG)n,有高度的多态性,分布位置恒定,是最常用的遗传学标记。几种卫星DNA的区别和功能见表3。此外,三核苷酸重复序列也较常见,此类重复序列过度扩增可引起遗传病,如脆性X综合征等。小卫星和微卫星DNA在基因组中的频率平均每2kb出现一次,占全基因组序列的3%。
散在重复序列(interspersed repeated sequence)分布散在,占全基因组序列的45%。根据序列长短分为短分散核元件(short interspersed nuclear element,SINE)和长分散核元件(long interspersed nuclear element,LINE)。SINE长度90~500bp,拷贝数106以上,平均2.2kb,分散于基因内、基因间或基因簇或内含子。人类基因组中最丰富的Alu序列是典型的代表,由282bp序列构成,约有50万~70万拷贝。LINE长度为5000~7000bp,重复拷贝数102~4次,如KpnI家族,这些序列构成转座元件,使DNA可在基因组内由一条染色体转移到另一条染色体上。
2.基因的组织结构
基因是有功能的DNA序列,包含编码序列和非编码序列。人的基因由外显子、内含子和侧翼序列组成,呈线状排列在染色体上。如图7所示。
表3 几种卫星DNA的区别和功能

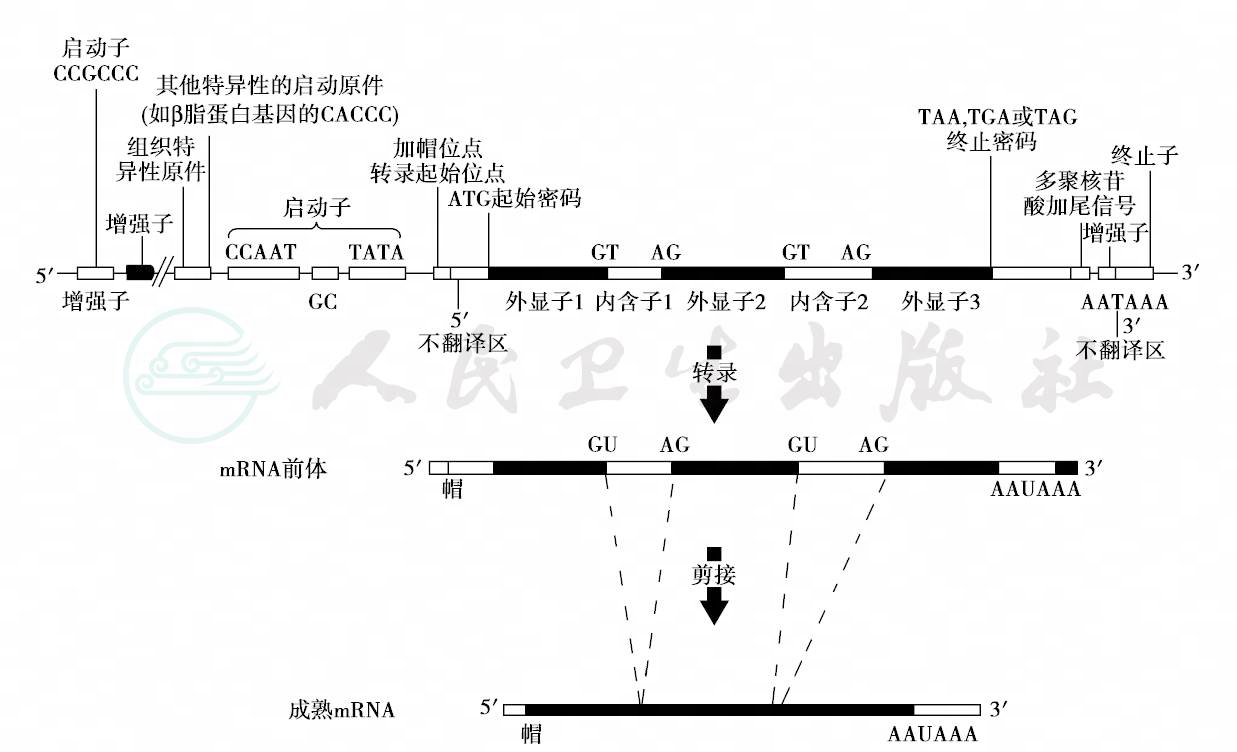
图7 基因的结构模式及转录模式
外显子(exon)多为编码序列;内含子(intron)是非编码序列,在转录为成mRNA之前被剪切掉。一般一个基因由若干个外显子和内含子组成,外显子平均长度小于200bp,内含子长度一般较长,平均长度为3000bp,没有内含子的基因较小,大多成簇存在,执行相似的功能。基因的大小可以相差很大。另外,外显子与内含子的关系并非完全固定不变,相同一段DNA序列,当它作为编码某一多肽链的基因时是外显子,而作为另一多肽链的基因则可成为内含子,所以,同一基因有转录为两种或两种以上mRNA的可能。
内含子与外显子相间排列,在每个内含子和外显子的接头区都有一段高度保守的共同序列,是RNA剪接信号。内含子5'端的两个核苷酸是GT,3'端是AG,转录时由5'端至3'端顺序将内含子剪掉,称为GT-AG法则。
侧翼序列是位于基因5'和3'端一段不转录的DNA序列,它们是基因转录的重要元件。5'端含启动子(promotor),一般位于基因起始点 ATG上游100~200bp间,包括在上游(-34~-36bp)的TATA框(TATA box)、CAAT框(CAAT box)和GC框。这些位于基因启动子中并且能够与转录因子特异性结合的保守DNA序列被称为顺式作用元件(cis-acting element),把转录因子(一些蛋白质)通常称为反式作用因子(trans-acting factor)。转录因子与启动子结合后激活RNA聚合酶,在特定位置启动基因转录。侧翼序列中还有增强子(enhancer)和沉默子(silencer),它们可以在基因的任何位置,增加或抑制基因的转录活性。侧翼序列的3'端由一段AATAAA和回文序列组成终止子(terminator),AATAAA是多聚腺苷酸(poly A)附加信号,回文序列转录后形成发夹结构,阻碍RNA聚合酶继续移动使转录终止。有些基因没有poly A,但在其上游有G/T簇或发夹式的高级结构,形成终止信号或终止子。基因的结构和转录模式见图7。
3.基因的生理功能
随基因组计划的进展,估计的基因数在不断地发生变化,体现了认识发展的过程。10多年前,多数科学家认为基因数约10万个。在2001年发表人类基因组工作草图过程中,这个数目估计只有3万~4万个,而人类基因组完成图的分析结果表明人体细胞含基因2万~2.5万个。
在人体每个细胞的2万~2.5万个基因中蕴含着构成和操控机体形成各器官的结构和功能的复杂信息。尽管每一个细胞含有相同的基因组,但在发育的不同阶段,不同的器官、组织以及在不同“环境”下基因的表达不同,以此构成器官和组织结构和功能的特异性。对基因组表达谱的研究能够帮助理解细胞和组织的生物学功能和阐明疾病的发生,有助于发现和研究基因功能,目前对其的认识还非常有限。神经系统是人类最复杂的系统,脑组织也是基因表达率最高的组织。
在遗传学上根据基因的功能,大致把基因分为两类:一类是结构基因,编码多肽链,经加工、修饰和形成各种高级结构后执行各种蛋白的功能,包括结构蛋白、酶、受体、各种转运蛋白等;还有一类是调控基因,不作为合成蛋白质的模板而只起调控基因表达的作用,包括启动子、增强子和沉默子等。这两类基因的功能异常均可导致人类疾病。
一些基因有几个启动子,经过选择性剪接使基因编码的蛋白数远远多于2.5万的基因数。另外,选择性剪接本身也具有重要的生物学功能,在正常和疾病组织中基因的剪接模式或不同,剪接本的比例有差异,提示这些差异与疾病有关。
4.基因复制(rep lication)
在细胞分裂过程中,DNA分子进行准确的复制,使分裂以后产生的子细胞获得相同的遗传信息。DNA复制是在细胞有丝分裂间期的S期中进行的,人类DNA复制的速率约每秒合成40~50核苷酸,相对较慢(细菌每秒合成500~1000个)。复制过程:①DNA双螺旋分子经过解旋酶和解链酶作用将碱基对之间的氢键断开,使两条多核苷酸链分开;②每一条多核苷酸链内侧面即有暴露出来的碱基,经DNA合成酶作用各自能与互补的核苷酸以T对A、C对G通过氢键连接;③新连接上的多核苷酸与原来的多核苷酸链形成新的螺旋,在DNA聚合酶的催化下,一个DNA分子就复制成两条结构完全相同的DNA分子。由于复制后的DNA分子是由一个新链与一个旧链构成的,所以称为“半保留”复制(semi-conservative replication),见图6。
DNA分子的两条链反向平行,一条5'→3',另一条3'→5'。DNA复制是从特异的复制点开始双向复制,复制方向按5'→3'进行,以3'→5'链为模板的5'→3'方向的链进行连续复制,速度较快,完成早,称为前导链(leading strand);以5'→3'链为模板的反向链,在按照5'→3'方向复制过程中,先合成100~1000bp的冈崎片段,然后由DNA连接酶将复制的片段连接起来形成一条完整的单链,这个复制过程速度较慢,称为后随链(lagging strand)。DNA复制中一条链连续,另一条链不连续的复制方式称为半不连续复制(semi-discontinuous replication)。
5.基因表达(gene exp ression)
基因表达是DNA序列蕴藏的遗传信息通过转录和翻译最终合成蛋白质的过程。
(1)转录(transcription)
系DNA分子上的遗传信息传递到信使核糖核酸(messenger ribonucleic acid,mRNA)的过程。开始转录时,RNA聚合酶Ⅱ结合到DNA的启动子,在其作用下中DNA双链打开,以DNA双链中的一条3'→5'单链(模板链或称反义链)为模板,按照碱基互补配对,RNA的U(Uridine,尿嘧啶)与DNA的A配对,以三磷酸核苷酸(NTP)为原料合成RNA前体。然后经加工修饰,包括:①加帽(capping):5'端加7-甲基鸟嘌呤核苷酸;②加尾(tailing):3'端加100~200个poly A尾;③剪切酶作用下按GT-AG法则剪切掉内含子,最后将外显子由连接酶连接成成熟的mRNA。见图7。mRNA通过核膜进入细胞质与核糖体(ribosome)附着。
(2)翻译(translation)
以mRNA为模板翻译成氨基酸序列的过程称翻译,这个过程是在细胞质的核糖体上进行的。各种蛋白质由20种氨基酸以不同方式和数目组合而成。这些氨基酸连接成多肽链,最后形成具有空间结构的蛋白质。
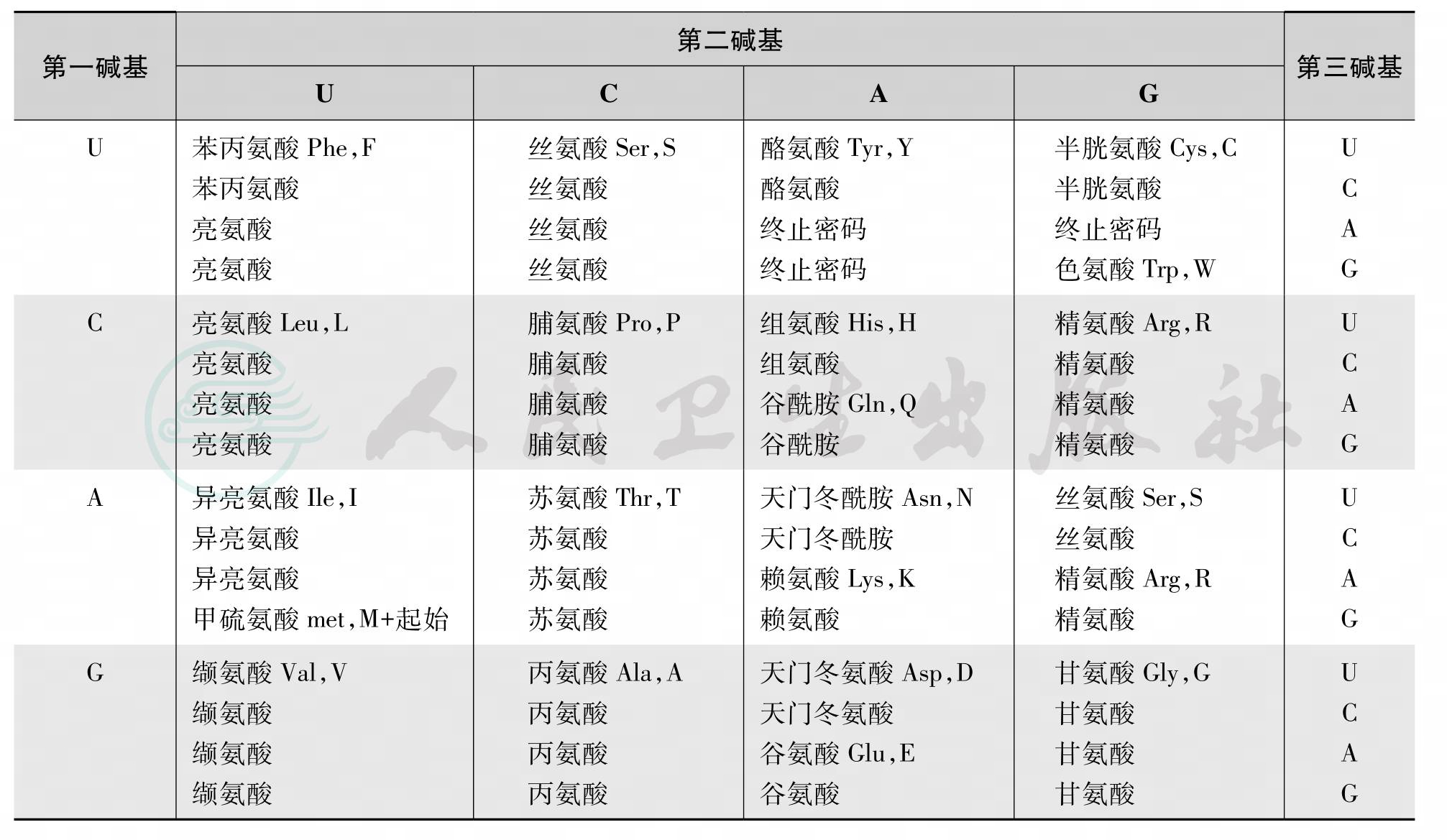
mRNA作为蛋白质合成的模板,以核苷酸序列的形式指导多肽链氨基酸序列的合成,从mRNA 5'端的起始密码到终止密码子前的一段序列代表一个基因,称为开放读框(open reading frame,ORF)。ORF内每三个相邻的碱基组成一个遗传密码(genetic code),又称三联密码子(triplet code)或密码子(codon),决定一种氨基酸。核苷酸分子有4种碱基,组成43=64个密码子。其中有61个密码子分别为20种氨基酸编码,其余3个不编码氨基酸,为蛋白质合成的终止信号,即终止密码子(stop codon)。AUG代表起始信号,也是甲硫氨基酸(蛋氨酸)的密码子,见表4。
表4 遗传密码表
多肽链的合成是在细胞质中的mRNA、tRNA(transfer ribonucleic acid,转移RNA)及核糖体协同作用下进行的。核糖体是蛋白-rRNA(ribosome RNA)复合物,由大小亚基(60S和40S)组成,小亚基识别mRNA 5'端的帽,合成从AUG开始,tRNA上的反密码子识别与mRNA互补的密码子,核糖体的大亚基结合小亚基开始精确地合成肽链,整个过程按进位、转肽、移位和脱落的步骤不断重复至终止密码子,多肽链从核糖体释放出来。翻译过程中常常是几个核糖体,被称为多聚核糖体(polysome),同时在一条mRNA分子上进行翻译(图8)。随之,mRNA与核糖体也就分开。翻译后的多肽链需要加工修饰,主要有脱甲酰基、乙酰化、磷酸化、糖基化、链切割等,以及两条及以上的多肽链间的连接或折叠形成一定的立体空间构象。细胞内合成的蛋白质经过靶向运输到达相应的功能部位,在细胞内外发挥生物活性作用。
DNA分子上的遗传信息通过转录为mRNA,再经过翻译的过程,最后合成蛋白质行使功能。DNA→RNA→蛋白质的信息传递原则就是遗传中心法则(genetic central dogma)。由于发现存在逆转录酶,能以RNA为模板合成DNA,因此,遗传信息也并非单一方向传递。

图8 DNA遗传信息的转录和翻译图解
6.基因表达的调控
机体每个体细胞中蕴藏着全套的遗传信息,但在特定的时间和空间只有部分基因表达,这是实现基因功能的基本保证。细胞类型的差别是由基因表达差异决定的。在个体发育过程中,一个受精卵经过一系列的细胞分裂和分化形成不同类型的细胞和组织,分化就是不同基因表达的结果,不同发育阶段和不同类型细胞的基因表达时空上接受严密的调控。当基因表达发生时空错误或表达产物的质和量异常时都可能导致疾病发生。
在多数细胞中都有表达的基因称位管家基因(housekeeping gene),如DNA复制、RNA转录、蛋白质合成的酶基因、控制糖酵解和三羧酸循环以及细胞骨架蛋白的相关基因等。细胞特异性表达的基因称为奢侈基因(luxury gene),如红细胞的血红蛋白、胰岛细胞的胰岛素和结缔组织的胶原蛋白和弹性蛋白等。
基因表达的调控是非常复杂的,调控的基本形式发生在DNA水平、转录水平、转录后的修饰、翻译水平和翻译后修饰及表观遗传学调控等多种不同层次,但最关键的调节仍然是转录水平。①DNA水平的调控是通过改变基因组有关基因的数量、结构顺序或活性来实现调控,包括基因扩增、丢失、重排和修饰。DNA甲基化也可使基因失活,甲基化多发生在CpG二核苷酸对。甲基化CpG序列与甲基化CpG结合蛋白结合后被转录抑制因子和组蛋白去乙酰化酶组成的复合体识别,使染色质形成紧密结构而无转录活性。组蛋白的乙酰化程度也是影响转录的一个重要因素,乙酰化的组蛋白对DNA的亲和力减低,使染色质松散易于基因表达。②在转录水平,有效的转录需要大约50种不同的蛋白质组成的复合物之间的相互作用。除位于基因启动子区的参与基因表达过程的顺式作用元件外,需要反式作用因子通过DNA-蛋白质相互作用特异性调控基因表达,如类固醇激素通过调节该基因启动子中激素反应元件与激素-激素受体复合物的特异性结合调控表达;转录因子之间也可以通过形成同源和异源多聚体参与调控组织特异性基因表达;特异基因的转录活性还能被远距这个基因数千个碱基的增强子所增强,增强子并非直接作用于这个基因,而是通过一个中间的活性因子与基因相互作用,与之相反,沉默子也可以远距离地抑制转录。③转录后的修饰,如差异性剪接:一个基因的转录本通过对转录起始点和终止点选择性剪接形成异构体,现在的基本观点认为90%的基因可能有使用不同外显子构成的异构体。异构体可能构成的功能从最大到相反多种层级,提高控制的准确性。④小RNA(microRNA,miRNA)是在真核生物中发现的一类内源性有调控功能的非编码RNA,大小为20~25bp,组成RNA诱导的沉默复合体的一部分。通过碱基互补配对识别靶向mRNA,可能通过影响mRNA的稳定性和蛋白质翻译水平而抑制表达,即主要参与基因转录后调节,有高度的保守性、时序性和组织特异性。目前对miRNA的生物学功能了解还不多,已知其参与调解细胞生长、组织分化,因而与生命过程中发育和疾病有关。
(三)基因组计划与基因组图谱
基因组是一个生命体遗传信息的总和。人单倍体基因组含3.2× 109bp的DNA序列,分布于22条常染色体和X、Y性染色体。基因组的信息在很大程度上决定了人的生长、发育、生殖、疾病、衰老和死亡的所有生命过程。因此,阐明基因组 DNA序列及其功能意义重大。2004年10月国际人类基因测序协作组(IHGSC)在Nature上公布了人类基因组的完成序列,它覆盖了99%的常染色质区,根据测序的结果推测人类基因组含2万~2.5万基因。
基因组作图有两种方式:遗传连锁图(genetic linkagemap)和物理图谱(physicalmap)。①遗传连锁图使用连锁分析的方法,计算连锁的遗传标记或基因之间的重组率确定相对距离,用厘摩(centimorgan,cM)即每次减数分裂的重组率为1%表示,1cM大约相当于物理图谱的1Mb。遗传连锁图用多态性的标记作为界标,包括早期第一代的限制性片段长度多态性标记(restriction fragment length polymorphisms,RFLPs)、第二代的微卫星标记或短串联重复(short tandem repeat,STR):重复长度 2~6bp,如(CA)n、(CAA)n、(AAAT)n等,含量丰富,重复数目变化大,检测方法易操作;第三代的单核苷酸多态性(single nucleotide polymorphisms,SNPs),基因组中估计有3×106~10×106,利用相邻的SNPs构成单体型(haplotype)还可有更多个基因型,提供更多的多态性信息。2002年10月启动了国际人类基因组单体型图谱(HapMap)计划,目标是构建人基因组DNA序列中多态位点的常见模式。用高通量的DNA芯片技术检测SNPs已成为识别和定位疾病基因的一种手段。②物理图谱:用染色体定位明确的单拷贝序列标签位点(sequence tagged site,STS)作为界标,并用以YAC(yeast artificial chromosome)或BAC(bacterial artificial chromosome)为载体构建的连续克隆,克隆含有覆盖每一条染色体的重叠片段DNA,称为叠连群图谱(conting map)。这是大规模DNA测序的基础,也是基因序列图的雏形。
基因组测序最直接的应用就是促进疾病相关基因的识别。遗传连锁分析是确定疾病相关基因最常用的方法。选择DNA多态性标记对受累家系进行连锁分析、初步定位与疾病相关的染色体区段,克隆该区的DNA片段,通过测序查找突变,这就是定位候选克隆的策略(图9)。利用这样的方法定位了很多单基因病的致病基因,如图所示的X染色体基因定位(图10)。这些信息可以通过互联网在免费的公共数据库查询。明确致病基因才有可能在未来实现基因治疗。
我国是人口大国,遗传资源丰富。历史、地理、经济、文化等使中华民族形成56个族群与众多隔离族群,近交系、大家系以及染色体变异引起的疾病,也有其特殊性,研究方向极为广阔,资源极为珍贵。HGP计划的完成对遗传病防治、改善环境、使人民达到健康长寿的目的将会指日可待。
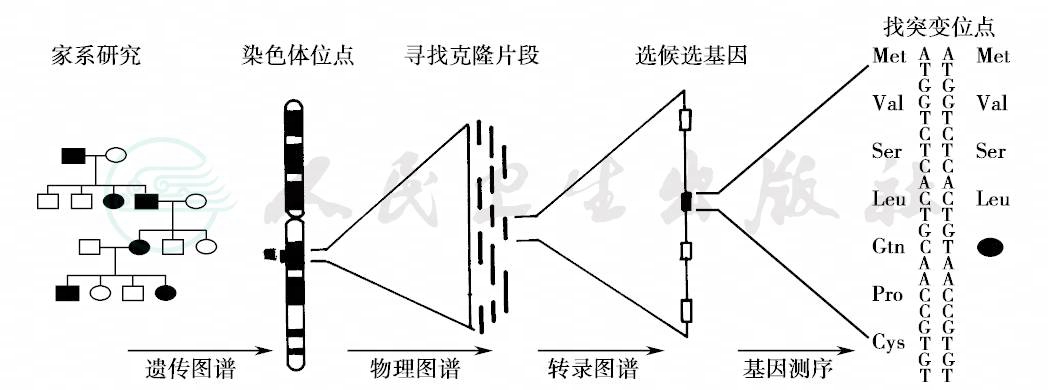
图9 基因图谱的研究过程——寻找遗传疾病基因的常见处理
图10 人类X染色体上定位的基因示意图
(四)遗传变异和基因突变
遗传变异源自基因组DNA突变(DNA mutation),突变是DNA序列中的碱基改变。DNA能够发生自发性突变,突变也是进化的动力。人类DNA的突变率约为百万分之一,大多数突变可以自发性修复,这是保持遗传信息稳定性的重要基础。一些突变导致疾病;一些突变未发现与疾病相关,构成人类基因的多态性。
1.基因突变
突变可以发生在编码序列或非编码序列,发生在生殖细胞而传递给子代,也可以发生在体细胞不传递给子代。有害的突变构成群体的遗传负荷,引起遗传性疾病,但是相对罕见。物理、化学和生物等因素能够损伤DNA而诱发突变;突变也与基因大小和序列有关,如人体中较大的基因DMD、血友病A和神经纤维瘤病Ⅰ型的基因有高突变率;CpG二核苷酸常常是DNA序列中的突变热点,在哺乳类80%的CpG被甲基化,甲基化的C容易脱NH2转变为T而突变,CpG突变率比其他序列高12倍,如Rett综合征的致病基因MECP2突变中常见的8个点突变均为C>T的突变;突变与父母生育年龄相关,一些常染色体显性遗传病,如马方综合征、软骨发育不全、神经纤维瘤病和Apert综合征随父亲年龄增大发病率增加,而母亲高龄更多与染色体病相关。常见的突变类型有单碱基替换(substitution)、缺失和插入(图11)。

图11 突变方式示意图
(1)点突变(pointmutation)
指DNA序列中的一个碱基被另一个碱基替换,最常见。嘧啶与嘧啶或嘌呤与嘌呤之间的替换称为转换(transition);嘌呤与嘧啶之间的替换称为颠换(transversion)。如果突变发生在非基因的DNA序列上,一般不产生效应;发生在基因的调控序列可能改变基因的表达水平;发生在编码序列可使mRNA的密码子发生改变,最终影响多肽链中氨基酸序列,出现下述不同的突变效应:①同义突变(same sense mutation),碱基替换后,三联密码子虽然改变了,但编码同一种氨基酸,因此,一般不影响蛋白质的功能。这种突变常发生在密码子的第三个碱基。②错义突变(missense mutation),碱基替换后使mRNA的密码子改变成编码另一个氨基酸的密码子,改变了氨基酸序列,常影响蛋白质功能。这种突变常发生在密码子的第一或第二个碱基。③无义突变(nonsense mutation),碱基替换后,使一个编码氨基酸的密码子变成了终止密码子(UAG、UAA或UGA),导致肽链合成提前终止,因肽链变短而失去功能。
(2)移码突变(frame shiftmutation)
指在DNA的编码序列中插入或丢失一个或几个碱基,使插入点或缺失点下游的DNA读码框发生改变,突变点以后的氨基酸序列都发生改变。
(3)DNA大片段突变
是相对上面两类的“小”,包括缺失(deletion)、插入(insertion)、重复(duplication)和 DNA重排(rearrangement)。缺失DNA的片段可以从数十至数万个碱基,源于DNA的重组,可以引起结构基因序列的丢失或编码序列重排而影响整个基因,使其功能丧失。插入的DNA片段可能源自同一条染色体或其他染色体或外源基因,对基因功能的影响取决于插入的位置,可能影响基因表达。缺失和插入均可产生DNA重排。重复突变指染色体某一区段DNA序列重复改变引起基因拷贝数增加或基因产物的剂量增加,与染色体的不等交换有关。
(4)动态突变(dynamic mutation)
基因组中短串联重复序列,特别是位于基因的编码序列或侧翼区的三核苷酸重复扩增是主要形式,如CGG、CAG或GCG等重复扩增。重复次数在上下代传递的过程中明显增加,导致遗传病的发生。除三核苷酸外还有五核苷酸和24核苷酸的小片段重复突变也可以发生在染色体水平。包括染色体数目和结构的改变,见染色体病。
2.多态性(polym orphism)
指两种或两种以上变异类型或基因型并存的现象。一般认为每种变异型的频率超过1%,小于1%称为罕见多态性。
(1)SNPs
是人类基因组中最多见的一种多态性改变,一个人平均大概有3×106个SNPs,说明个体间每千个碱基中就有一个不同。有两种SNPs的生物学意义可能更大:其一位于基因的编码区的SNPs称为cSNP,若引起蛋白重要氨基酸的改变可能影响功能;其二位于调控区的SNPs可能影响表达量。目前认为这两种SNPs是决定人类表型多样性的核心信息。
(2)拷贝数变异(copy number variants,CNVs)
指人群中存在的大于1000bp的DNA片段,可以有多个拷贝,每个体间平均大概有4Mb不同。一些CNVs已经证明与遗传病相关。
多态性主要影响复杂性或常见病的患病风险,如唇腭裂、先天性心脏病、糖尿病、高血压、冠心病等。
3.突变原因自发性或诱发性
已知某些物理辐射线(如X线、α线、γ线、β线、中子射线和紫外线)、化学物质(如氮芥、环氧化合物、吖啶黄、亚硝酸、甲醛、烟草中尼古丁等毒物等)、病毒感染以及温度改变等均可诱发突变。发生自发突变的因素还不清楚,可能与天然辐射线有关。
(五)遗传的基本规律
生物体中各个遗传性状受其相应的基因控制。体细胞基因成对(一对等位基因),在形成配子时彼此相互分离。所以,一个配子中只有一对等位基因中的一个基因,这是孟德尔遗传定律中的分离律。等位基因的性状分为显性和隐性,显性基因只需要一对等位基因中有一个存在,就能够得到表现。例如L代表高身材的显性基因,具有一个L基因的个体就是高个子。高大的身材是可以观察到的性状,称为表现型或表型(phenotype)。决定这个表型的基因形式称为基因型(genotype)。对高个个体而言基因型可以有一个L基因伴一个矮基因或伴一个正常个头基因,也或两个均是L基因,他们表型相同,而基因型则不同。隐性基因只有一对等位基因都存在时才能够得到表现。例如a代表白化病的隐性基因,如果一个a基因与一个代表正常皮肤颜色的显性基因A配对时,就不会表现白化病,只在基因型为aa时才表现白化病。aa或AA两种基因型有两个相同的隐性或显性基因称为纯合子(homozygote)。Aa有一个显性和一个隐性基因称为杂合子(heterozygote)。不是同源染色体的两对以上的基因,在形成配子时每对等位基因可相互分离,在形成合子时还能随机地自由组合,各不相扰,这是孟德尔遗传定律中的自由组合律。一条染色体上有很多个基因,它们在染色体上彼此之间是连锁在一起的,构成连锁群;同源染色体上的基因连锁群并非固定不变。在生殖细胞形成过程中,同源染色体配对联会、发生交换使连锁群发生重新组合,这就是连锁和互换律。同源染色体上的两对等位基因之间发生交换与基因间的距离有关,相距越远,发生交换的几率越大。
分离律、自由组合律和连锁与互换称为遗传学的三大定律,也是遗传的基本规律。
(六)遗传与环境的关系
机体不能脱离环境而孤立地生活,环境的变化也必然要影响机体的代谢和生长发育,生物体的一切性状都是基因组信息和环境相互作用的产物,即基因型+环境产生表现型。环境包括内环境(母体的生理、病理或解剖异常)和外环境(感染、药物、饮食、辐射等)均可致基因突变或染色体畸变,形成出生缺陷等,如代谢性疾病或先天畸形。一种基因型在不同的环境条件作用下所能形成的遗传性状和疾病表现程度和特征可有不同,它的全部表现型,称为反应规范(reaction norm)。有的基因型所决定的反应规范比较狭窄,例如常染色体隐性遗传的白化病患者,基因型为突变的纯合子(或杂合子复合),不论在任何环境下,都不会产生黑色素,表型就是白化病。但也有反应规范比较宽广,在不同的环境条件下,可以出现不同的表型。例如常染色体显性遗传的先天性成骨不全患者当中,表现度(expressivity)不同,有的表现轻微,仅表现蓝色巩膜;有的表现严重,除蓝色巩膜外还表现有耳聋、不同程度的骨折及牙本质发育不全等。这可能是同一基因型在不同的内环境(包括基因环境)中发展的结果。
当一种基因型在一定的环境中发育形成相应的表型,这种基因的作用得以外显。但也有随着环境(包括基因环境)的变化,基因型不能形成相应的表型,使外显不完全。在常染色体显性遗传中,有时会发生间隔一代的现象。更为明显的是,环境因素对某些遗传性疾病的发病起着重要的作用。例如半乳糖血症、苯丙酮尿症、对伯氨喹啉敏感者、线粒体基因1555G>A致耳聋等先天性代谢缺陷的临床表现是否出现,与饮食成分或服用某些药物有着密切的关系。对这些患儿,目前虽不能改变其携带的致病基因,但如能及时发现,防止与有害的环境因素(如半乳糖、苯丙氨酸、伯氨喹啉和使用氨基糖苷类药物)接触,可使疾病不发生或得到控制。
环境因素致畸最敏感的时期是在胚胎前3个月,此时为神经管闭合,脑、心、肾发育,肠转位,四肢生长,颜面愈合等的关键时期,可引起许多大器官的畸形和功能异常,以后则影响较小。致畸物剂量的大小也有关系,一般认为大剂量可以致死,中等剂量可以致畸,小剂量可能只引起发育迟缓。
从病因上看,所有的疾病都在一定程度上是由遗传所控制的,但另一方面也可由环境造成。而遗传与环境两个因素,在不同的疾病之中,其重要性也相应不同(图12)。
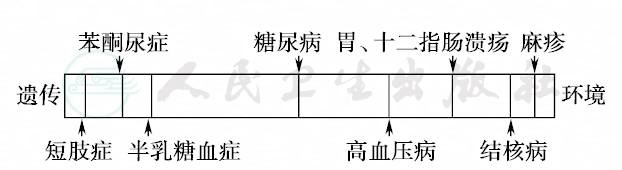
图12 遗传因素与环境因素在发病中的相对重要性示意图
图12中说明靠近遗传的一端是单纯的遗传性疾病。如短肢症,由一个显性突变基因所控制,苯丙酮尿症和半乳糖血症在隐性纯合子(或杂合子复合)中出现表型。这两种病已稍离开“遗传”的一端,因为环境因素(如食物)可以引起患儿表型的改变。在环境的一端是传染病,但同样不在最端部,因为在发病季节里,同样两个人接触病原体,一个发病,另一个可以不发病,这是由于每人遗传背景对疾病的易感性不同的关系。糖尿病、高血压病等疾病的遗传因素与环境因素对表型都起着几乎同等的重要作用。
不同类别遗传病的传递方式(mode of transmission of hereditary diseases)不同。
(一)单基因遗传病
单基因遗传病或称单基因病(monogenic disease,single gene disorder)指由一对等位基因控制而发生的遗传病,这对基因称为主基因(major gene),单基因病在上下代之间的传递遵循孟德尔遗传定律。根据主基因在的染色体的定位和等位基因的显性与隐性特征分为:常染色体遗传,包括常染色体显性和常染色体隐性遗传;性染色体遗传,包括X-连锁显性和X-连锁隐性遗传及Y连锁遗传。
单基因病的传递方式从系谱(pedigree)着手,即通过患者(先证者,proband)追溯其家族成员,包括直系和旁系亲属的数目、亲属关系和某种遗传病或表型的分布,按一定的格式绘制家系图。完整的系谱图既要有患者也要有正常成员。家系图常用符号及代表意义如下图13。
1.常染色体显性遗传(autosom al dom inant inheritance,AD)
致病基因位于1~22号常染色体上,呈显性性状,以A表示,其相应的另一个正常等位基因(野生型)用a表示。在完全显性的情况下,杂合子(Aa)与显性纯合子AA表型相同。由于显性致病基因在群体中的频率很低,因而实际上绝大多数显性病患者的基因型为Aa。常见有:多指(趾)、并指(趾)、短指(趾)、多发性外生骨疣、先天性成骨不全、Apert综合征、α型地中海贫血、遗传性出血性毛细血管扩张症、神经纤维瘤病、结节硬化症、成人型多囊肾等疾病呈显性方式传递(部分见表5)。AD的特点如下:①致病基因位于常染色体,因而与性别无关,男女患病几率均等。②患者双亲有一个患者,致病基因由亲代传递给子代,子代出现与亲代相同表型,即呈现垂直传递。由于绝大多数为杂合子,患者的同胞有50%的患病风险。③系谱中通常连续几代可以看到患者,即存在连续传递。④双亲无病时,子代一般不会患病,如患病可能为新生突变(图14、图15)。
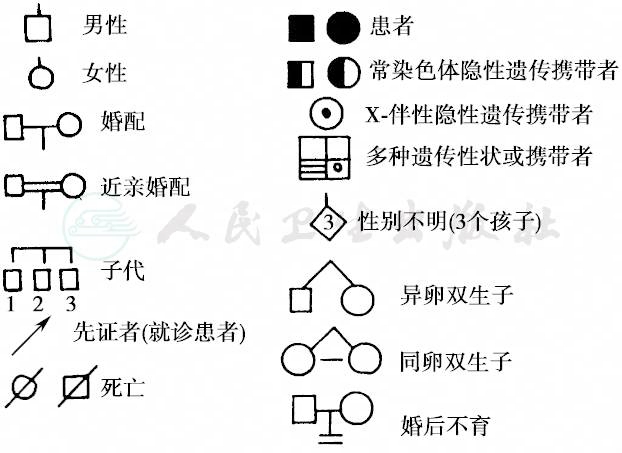
图13 家系调查中常用符号
由于基因突变及表达受多种复杂因素的影响,AD还有特殊的遗传现象。①完全显性:AD杂合子基因型Aa表现与纯合子基因型AA表型完全相同。如Huntington舞蹈症,占AD的少数。②不完全显性或半显性遗传(incomplete dominance or semidominant inheritance):AD杂合子Aa与纯合子AA的表型严重程度不同,即不完全显性或半显性遗传(incomplete dominance or semidominant inheritance),多数AD基因的表达属于不完全显性。例如成骨发育不全,纯合子AA个体病情严重,常于婴儿期死亡;杂合子Aa病情较轻,可以仅表现蓝色巩膜。③共显性:一对等位基因在杂合子时,无显隐性之分,两种基因的作用都表现出来,称为共显性遗传(codominant inheritance)。人类的ABO血型的遗传就属于这种方式。ABO血型决定于一组复等位基因,即:IA,IB,i,可以构成6种基因型。但是IA和IB同时存在时,可以共同表现出来,称共显性,而IA和IB对i都是显性,所以6种基因型只显示4种表型,即A、B、AB和O型。即是基因型为IA IA和IA i均为A型,IB IB和IB i为B型,IA IB为AB型,ii为O型。④延迟显性:AD杂合子Aa致病基因在早期不表达,到一定年龄后才表达致病称为延迟显性(delayed dominance),如Huntington病的杂合子35岁后发病。⑤不规则显性(irregular dominance):AD杂合子Aa的显性基因由于某种原因不表现出相应的表型。换而言之,在一个家族中不是所有携带相同AD基因突变的成员均出现表型,但这个突变仍然可以传递给下一代并可能使其出现表型。这种显性最常见的原因是受外显率的影响。外显率(penetrance)指在群体有致病基因的个体中表现出相应表型的人数百分率,用“%”表示,在系谱中由于外显不全呈现隔代遗传现象。如多指症外显率75%。另外一个重要原因是受表现度(expressivity)的影响,在有表型异常的人中,表现的程度有的严重,有的较轻。表现度可能与修饰基因存在或环境有关。外显率与表现度是不同的两个概念,外显率代表基因表达与否,是“质”;表现度则是在表达的前提下表现出的程度,是“量”。
表5 一些常染色体显性遗传病的举例

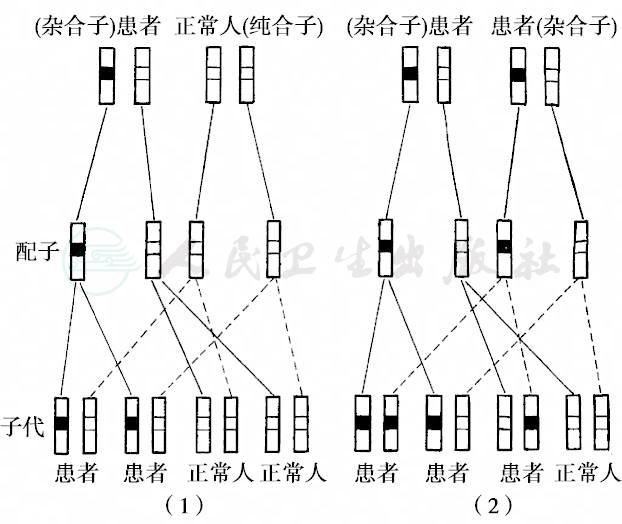
图14 常染色体显性遗传
(1)仅一亲代带有致病基因;(2)双亲皆带有致病基因

图15 先天性成骨不全的谱系图
常染色体显性遗传
根据AD以上的遗传特点预测其再发风险:①父母一方患病,基因型Aa,其子女有50%的患病风险;②父母双方均患相同疾病,基因型均为Aa,其子女有75%的患病风险;③如果家族中父母的同胞或祖父母之一患病,而父母不患病时,子女一般无病,但受不完全外显的影响其子女可有一定风险,尽管患病几率较小。
2.常染色体隐性遗传(autosomal recessive inheritance,AR)
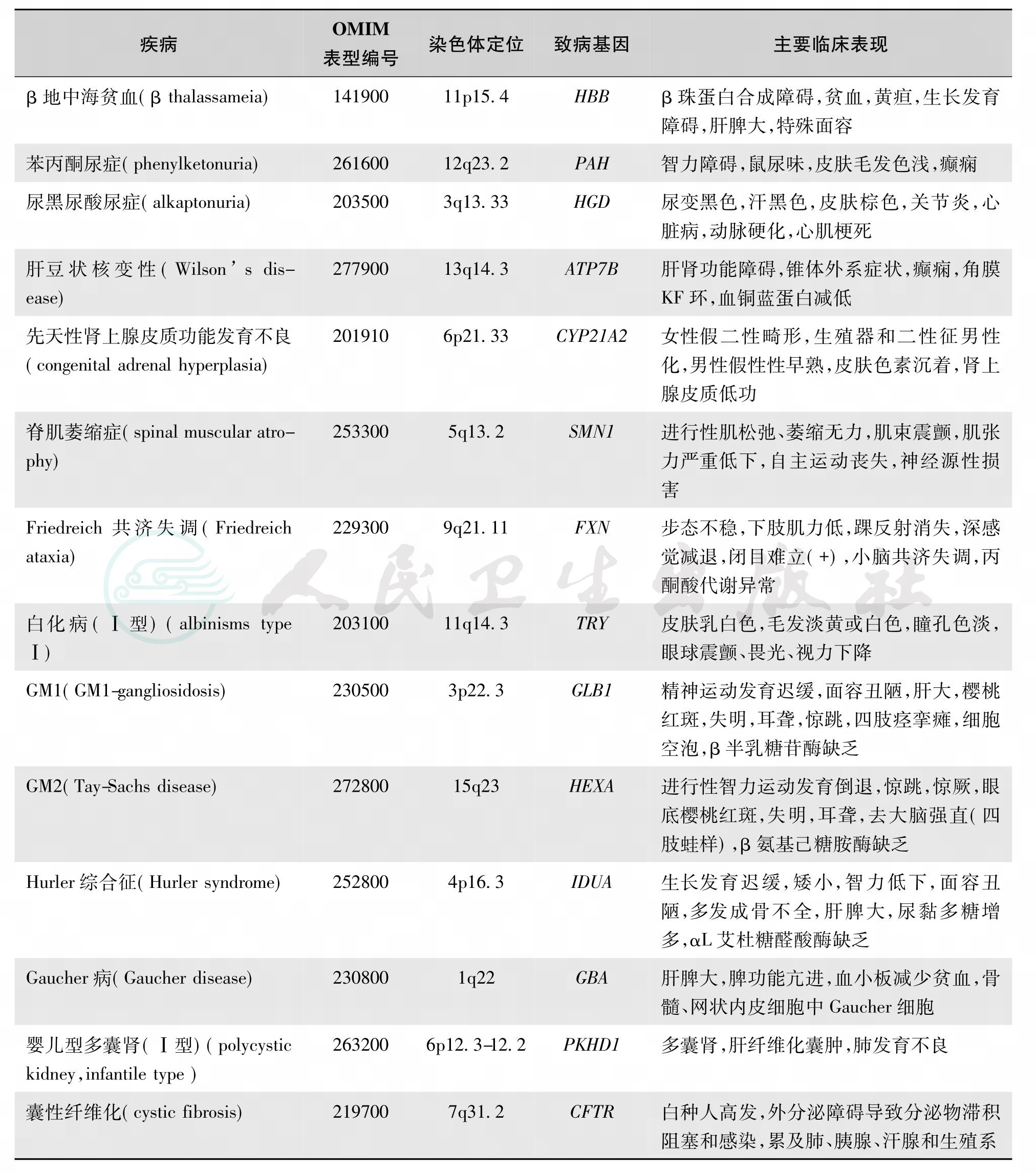
致病基因位于1~22号常染色体上,呈隐性性状。一对等位基因均存在突变,即纯合子或复合杂合子(compound heterozygous)才能出现表型,复合杂合子指一对等位基因突变位点不同。带有一个致病基因的杂合子个体,无异常表型,但能将致病基因传给子代,称为携带者(carrier)。故只有双亲都是携带者时,才有患者出现的可能性。AR的特点如下:①致病基因位于常染色体,因而与性别无关,男女患病几率均等。②患者在系谱中呈现水平分布,即患者在同胞中出现;患者父母(亲代)或子代不发病,患者在系谱中散发或隔代出现。③父母均系基因突变的携带者,同胞患病风险25%,携带者50%。④近亲结婚时后代风险明显增大(图16、图17)。临床上大多数的代谢性疾病为AR,如苯丙酮尿症和甲基丙二酸尿症。此外,白化病、肝豆状核变性、脊肌萎缩症、囊性纤维化、Crigler-Najjar综合征、范科尼综合征、半乳糖血症、糖原代谢病等约种属常染色体隐性遗传病(部分见表6)。
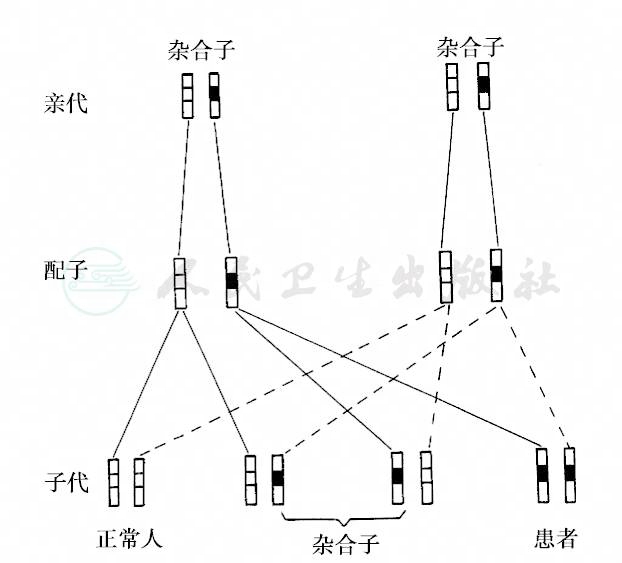
图16 常染色体隐性遗传
双亲皆为携带者
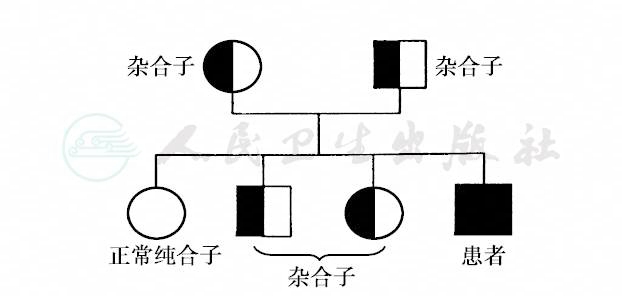
表6 一些常染色体隐性遗传病的举例
3.性染色体遗传的X连锁显性遗传(X-linked dom inant,X-LD)
性染色体遗传是指致病基因定位于性染色体。X连锁遗传与常染色体遗传相比,有以下特点:①男性为半合子(hemizygote),因为男性只有一条X染色体,等位基因数目相当于正常女性的一半,所以位于X染色体上的致病基因无论显性或隐性均表达而致病;②交叉遗传(crisscross inheritance),男性X连锁的基因只能来自母亲并传给女儿,无男性传男性;③女性杂合子表达有差异,一方面与基因的表达特性有关,另一方面与X染色体的随机失活有关。
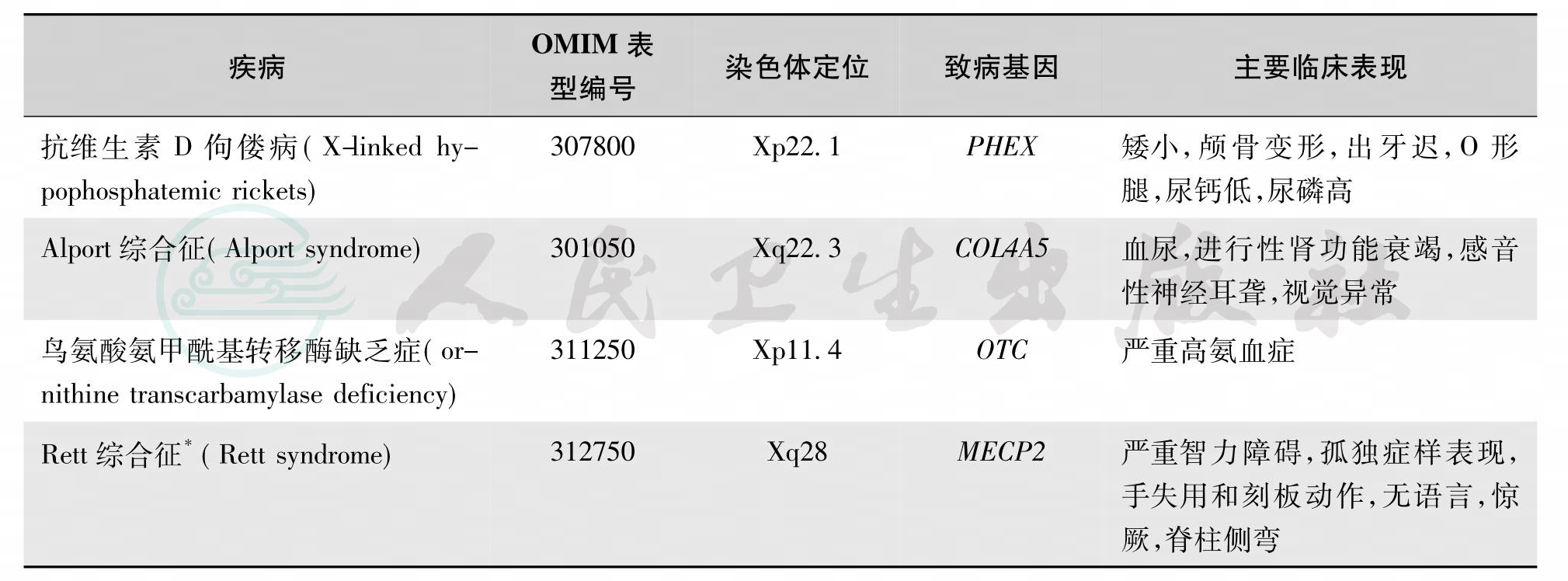
X-LD的特点如下:①患病群体中女性患者比男性多近一倍,相对女性病情较轻;②患者的双亲有患者;③男性患者的女儿均为患者,儿子正常;④女性杂合子的子女患病风险为50%;⑤系谱中有连续传递的现象(图18、图19)。这类遗传病比较少见,最典型的例子是抗维生素D性佝偻病(vitamin D-resistant rickets),又称X-连锁低磷酸盐血症性佝偻病(X-linked hypophosphatemic rickets),其他例子见表7。
表7 一些X连锁的显性遗传病的举例
注:*目前认为X-连锁显性,男性胚胎致死
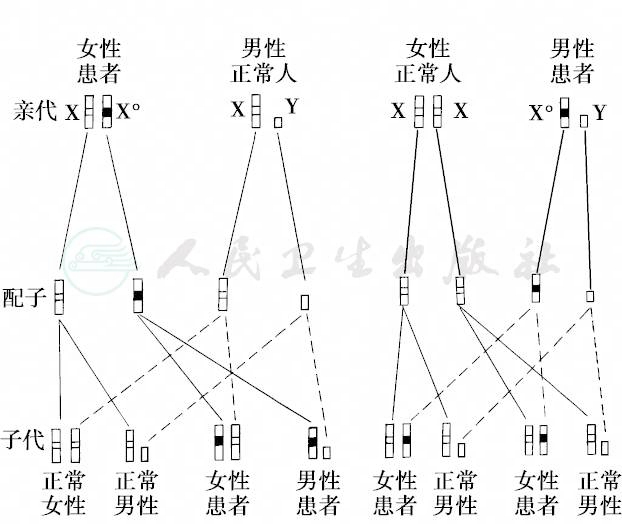
图18 显性伴性遗传
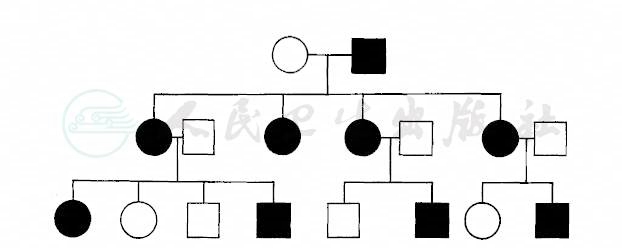
图19 慢性遗传性肾炎显性伴性遗传的谱系
(第二代子女中,女儿都发病)
4.性染色体遗传的X连锁隐性遗传(X-linked recessive,X-LR)
X-LR的特点如下:①患病群体中男性患者远比女性多,系谱中往往只有男性患者。②双亲没有患病时,儿子可能患病,女儿则不患病;儿子如果患病,母亲是携带者,女儿有50%的可能性为携带者。③男性患者兄弟、外祖父、姨表兄弟、外甥及外孙可能为患者。④如女性是患者,其父亲一定是患者,母亲则为携带者(图20、图21)。由于在群体中带有相同致病基因的个体进行婚配的机会甚少,在临床上极为少见。如Duchenne型性肌营养不良(Duchenne muscular dystrophy,DMD)男性患者,常常于4~5岁发病,20岁已失去独立生活的能力或死亡,故女性发病的可能性极小(涉及其他机制,如累及基因位点的相互易位,X染色体非随机失活)。血友病主要见于男性,偶可见于女性。部分的见下表8。
5.Y连锁遗传
致病基因位于Y染色体,随男性传递,女性不患病(图22)。Y遗传的遗传性状和疾病少,主要有睾丸决定因子(SRY基因)、H-Y抗原、外耳道多毛等。
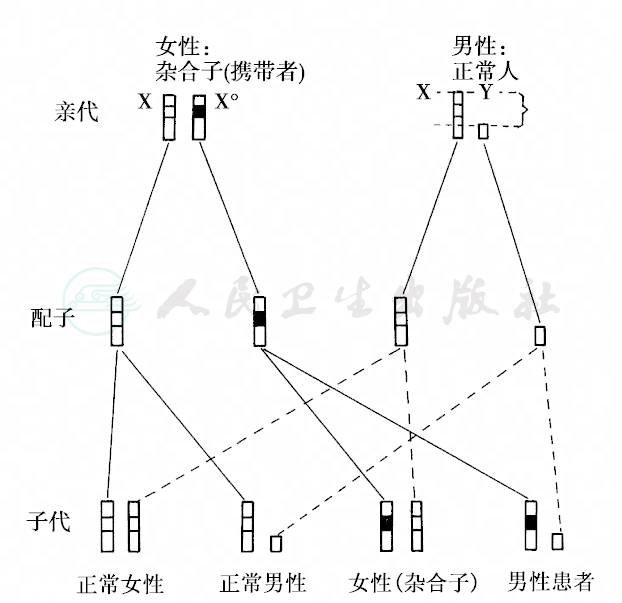
图20 隐性伴性遗传
X°代表位于X染色体上的病理基因;X代表位于X染色体上的正常基因
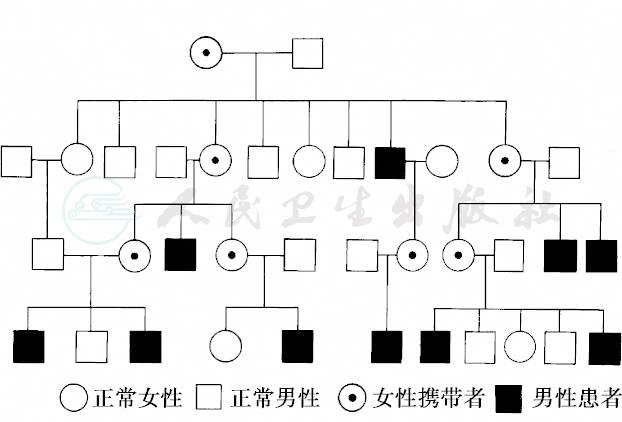
图21 血友病伴性隐性遗传的谱系图
表8 一些X连锁的隐性遗传病的举例
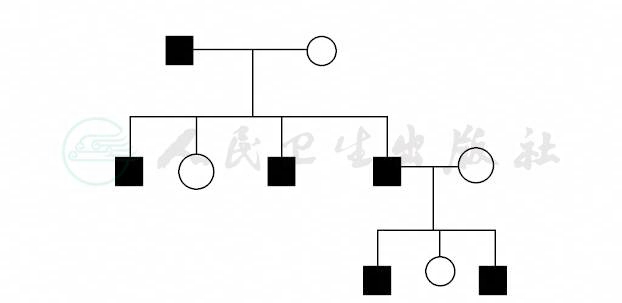
图22 Y连锁遗传
(二)多基因遗传
一些遗传性状或遗传病是由两对以上的多个基因共同作用的结果,彼此之间没有显隐性,是共显性的。每对基因的作用微小,故称微效基因(minor gene)。但是各对基因有累积的效应,累积后可以形成明显的表型效应。这些基因称为累加基因(additive genes),这样的遗传方式称为多基因遗传或多因子遗传(polygenic inheritance or multifactorial inheritance)。多基因遗传的性状在群体中的分布是连续的,有一个峰,即平均值。不同个体间的差异是量的变异,所以又称数量性状(quantitative character),以此相对于单基因遗传的质量性状(qualitative character)。由于多基因遗传病的发生还受环境因子的影响,因此,也称复杂疾病(complex disease)。
临床上一些先天畸形和疾病有一定的家族倾向,但是在同胞中的发病率远比单基因遗传的发病率为低,大约为1%~10%。这类遗传病为多基因遗传病。多基因病的形成受遗传和环境的双重影响,其中遗传因素所产生的影响程度称为遗传度(heritability),一般用“%”来表示。遗传度愈大,遗传因素对疾病的贡献越大,环境因素的作用越小;相反,一种多基因遗传病受环境因素的影响越大,遗传度越低。例如常见出生缺陷中的唇腭裂、先天性幽门狭窄和先天性髋关节脱位,幼年型糖尿病,支气管哮喘和精神分裂症等疾病,遗传度为70%~80%;无脑儿、脊柱裂、马蹄内翻足、冠心病和原发性高血压,遗传度为60%~70%;各型先天性心脏病、成年型糖尿病、消化性溃疡等的遗传度则不足40%。
在多基因遗传病中,遗传因素的作用只决定一个个体是否易于患某种遗传病的可能性,称为易患性(liability)。一般群体中易患性很高或很低的个体都很少,大部分接近平均值,故易患性是呈正态分布的。易感性(susceptibility)特指遗传因素决定的患病风险(个体所含有的遗传因素)。易患性决定多基因病的发病的最低限度称为发病阈值(threshold)。在一般群体和患者第一级亲属中,当这种易患性达到一定阈值即可发病。
多基因病的再发风险可按以下几种方法估计:①在一级亲属中可按Edward公式计算,即复发风险率(f)=群体发病率(P)‰,如唇裂在我国人群中患病率为1.7‰,则患者的一级亲属发病风险为1.7‰=0.4‰。此法对群体患病率高于10%或低于0.1%者则不够准确。②直接查阅各种比较常见的多基因疾病的再发风险值,如表9。③对表33-9中未列疾病,或已知某一疾病的遗传率、群体发病率或双亲与同胞中发病数,可由表10查出再发风险率。④已知群体患病率(%)、患者一级亲属患病率(%)或遗传度(%)三者之二,可按图23检索出另一项目。以上四种方法均可参考使用。
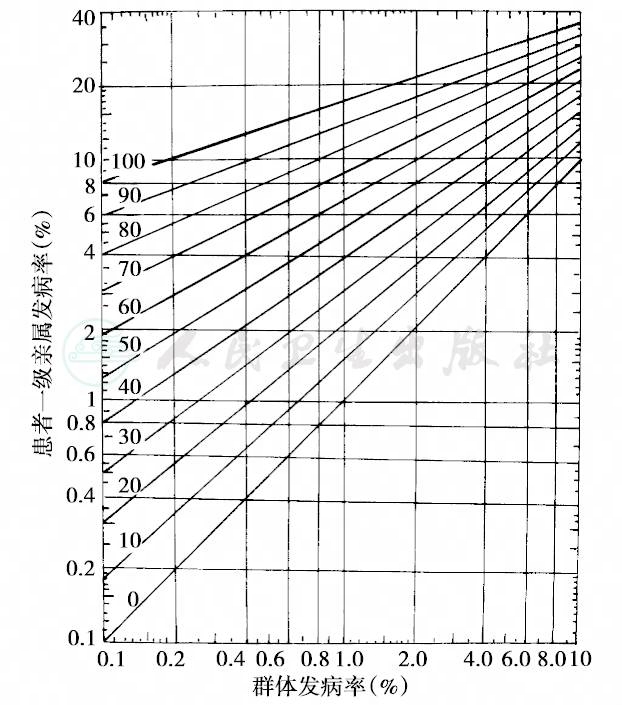
图23 群体发病率、易患性遗传度与患者亲属发病率的关系
斜线代表遗传度(%)
表9 常见多基因病群体患病率、先证者一级亲属患病率、性别比和遗传度(%)
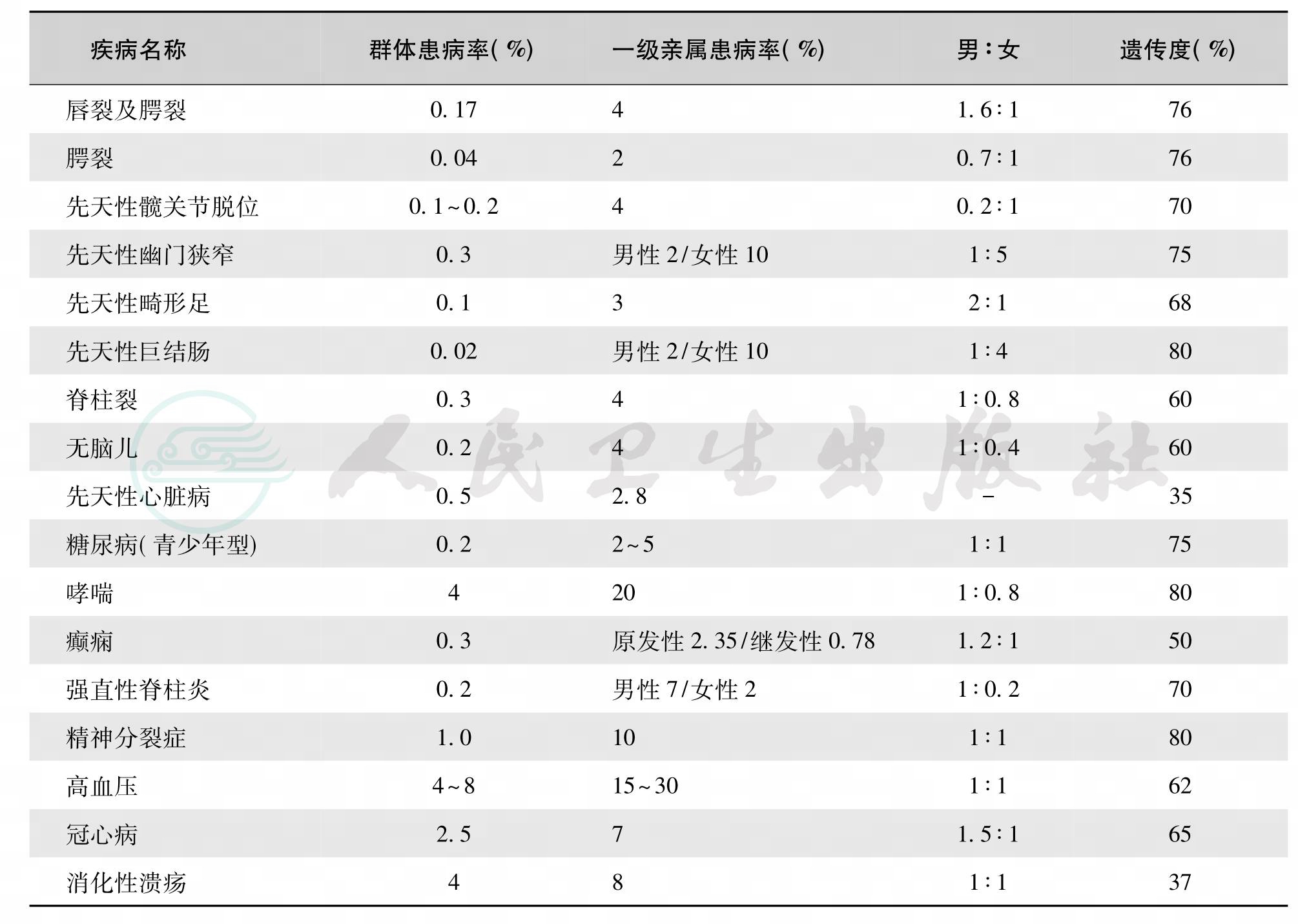
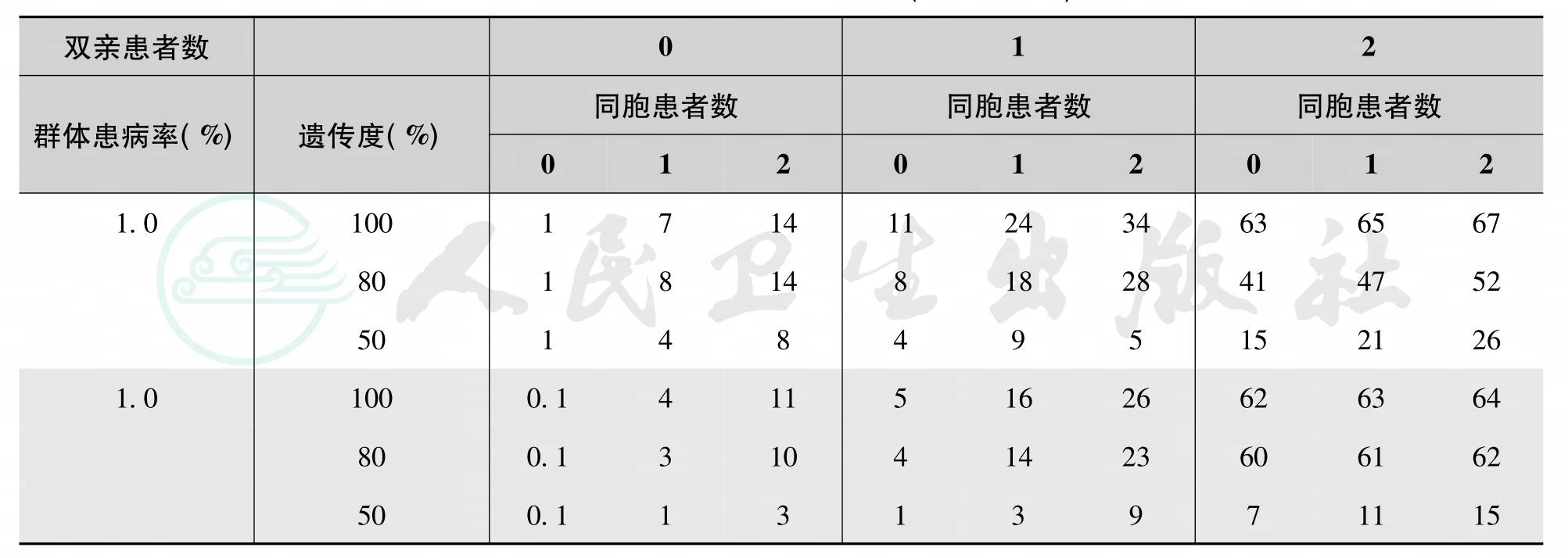
表10 多基因病再发风险估计(Sm ith表格)
综上所述,对多基因遗传可基本做如下估计:①在患者一级亲属(父母、同胞兄弟、子女)中的再发风险率与同代相近,大约为2%~10%。但偶尔在二级亲属或远亲中再发风险率可以偏高。②再发风险率与群体患病率有一定关联。③有些疾病的再发风险有性别差异,如先天性幽门狭窄,男性明显多于女性;先天性巨结肠,男性多于女性;先天性髋关节脱位,女性多于男性等。④同卵双生子患相同疾病的可能性为21%~63%,虽然不能达到100%,但远远超出孟德尔显性遗传的规律,因他们基因型相同。⑤同一家系中患病率高者,再发风险率也较高,这也不符合孟德尔遗传规律,如一对父母有一例单侧唇裂及腭裂的孩子,再发风险为4%,有两例者则可增至10%。⑥病情严重者的再发风险率高于轻度患者,如先天性巨结肠受累长度较长者较受累长度短者再发风险为高。
对多基因遗传病的再发风险估计还有其他方法,如连锁分析(linkage analysis)、受累同胞对分析(affected sib pair,ASP)、受累家系成员分析(affected pedigreemember,APM)、关联研究和连锁不平衡(associated studies and linkage disequilibrium)以及动物模型多基因分析,大多为研究性质,故不在此赘述。
(三)线粒体基因的遗传
线粒体位于细胞质中。线粒体是能量代谢的中心,细胞呼吸作用中的氧化还原反应在线粒体中进行,在这个过程中产生大量ATP,为细胞提供各种生命活动所需要的能量。线粒体DNA(mitochondrial DNA,mtDNA)是独立于细胞核染色体DNA外的位于胞质线粒体中的又一个基因组。
1.线粒体基因
人类mtDNA全长16 569bp:双链闭环分子,外环重链(H),G含量高;内环轻链(L),C含量高。包含37个编码基因,分别编码13个多肽链、22个tRNA和2个rRNA。重链分布28个编码基因,轻链分布9个编码基因。基因连续编码,没有内含子。有一个1122bp的D环,不编码,含有重链复制起始点、轻、重链转录启动子和4个高度保守序列,具有转录和调控功能,如图24。mtDNA的复制调控机制并未完全清楚。mtDNA有两个复制起点,非同步复制。复制时,由重链复制起始点开始,在DNA聚合酶γ的作用下单向进行,新合成的重链越过轻链的起始点后,轻链开始单向复制。因此,在轻链开始复制前,mtDNA有三股链组成,这是产生mtDNA大片段缺失的主要原因。mtDNA的转录则为双向同时进行,转录为多顺反子,即同一链上所有编码区都被转录,然后经过修饰和剪接形成成熟的RNA。一个线粒体内有2~10余个拷贝的mtDNA,一个细胞内通常有一百至数百个线粒体,故每一个细胞含数百至数千个mtDNA。线粒体DNA的转录可如图25所示。
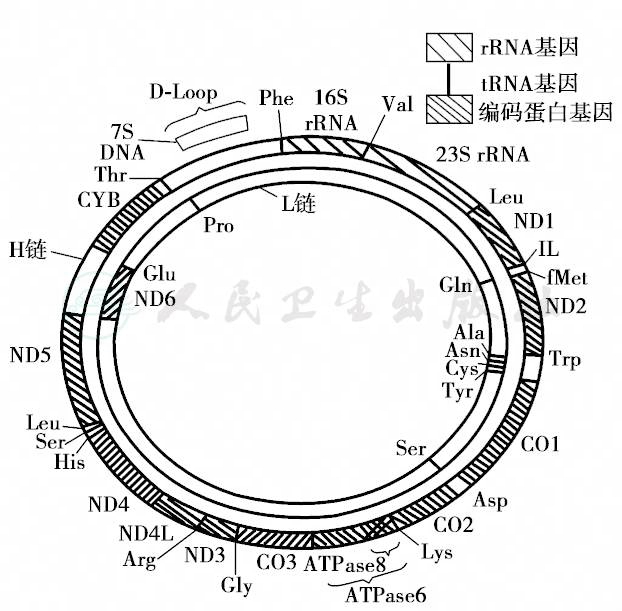
图24 人类线粒体基因组模式图
ND1-ND6:基因编码 NADH脱氢酶亚单位 1-6;CO1-CO3:基因编码细胞色素C亚单位1~3;Cyb:基因编码细胞色素B;Leu:亮氨酸;IL:异亮氨酸;Met:蛋氨酸; Trp:色氨酸;Lys:赖氨酸;Asp:门冬氨酸;Gly:甘氨酸; His:组氨酸;Ser:丝氨酸;Thr:苏氨酸;Val:缬氨酸; Ala:丙氨酸;Agn:门冬酰胺;Cys:胱氨酸;Tyr:酪氨酸; Glu:谷氨酸;Pro:脯氨酸;Arg:精氨酸;Cys:胱氨酸; ATPase:ATP酶:L链:轻链;H链:重链
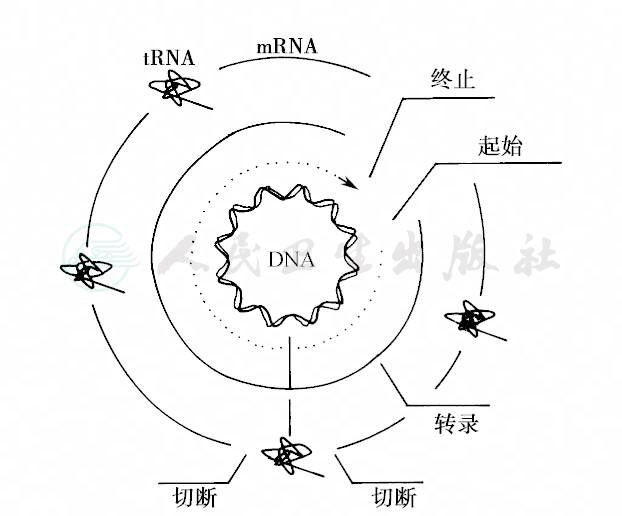
图25 人类线粒体DNA的转录
先转录成单一转录物,再由此切断并释出tRNA和其他RNA(引自Lewinb,1997)
2.线粒体DNA遗传特点
线粒体具有特殊的一些遗传特征。①在受精卵中,线粒体来自卵细胞,因此,mtDNA表现为母系遗传(maternal inheritance)。②不均等的有丝分裂:mtDNA在减数分裂和有丝分裂过程中要经过复制分离,卵母细胞中的极小部分mtDNA随机进入成熟卵子,使得亲代与子代之间可能产生差异;胚胎再经过细胞的有丝分裂和线粒体的自我复制使mtDNA数目增多,在这个过程中mtDNA仍然随机进入子细胞。如果母亲携带有突变的mtDNA,经过上述的细胞分裂过程,最终可能使子代的不同细胞及不同组织在发育的不同时期mtDNA突变比例不同。③同质性、异质性和阈值效应:如果同一个细胞的每一个mtDNA都相同,或全突变或全为野生型,称为同质性(homoplasmy);如果同一个细胞的mtDNA既有突变型,又有野生型,称为异质性(heteroplasmy)。异质性的细胞中突变mtDNA达到一定比例出现线粒体功能障碍称为阈值效应。阈值效应依赖受累细胞或组织对能量的需求,因此,高能量需求的脑、骨骼肌、心脏和肝脏等易受影响。④mtDNA突变率高,比核基因高10~20倍。与以下因素有关:mtDNA缺乏组蛋白与内含子形成的保护;突变后缺乏有效的修复系统;复制不对称,出现的单链DNA易发生自发性脱氨基而引起点突变;mtDNA处于高超氧化物环境更易损伤。⑤mtDNA突变可以在体细胞中积累,随年龄增长发展为均质性突变。
3.线粒体DNA突变
mtDNA突变的类型主要包括以下几类:①点突变:发生在蛋白质的编码序列上引起错义突变,如Leber遗传性视神经病(Leber hereditary optic neuropathy,LHON);发生在蛋白质翻译有关的tRNA或rRNA基因上影响mtDNA编码的全部肽链的翻译过程,导致呼吸链中多种酶合成障碍。典型的包括肌阵挛性癫痫伴破碎红纤维综合征(MERRF)、线粒体脑病-乳酸酸中毒-卒中样发作综合征(MELAS)等。②缺失或插入,缺失多见。常无家族史。缺失的片段较大,如常见的Kearns-Sayre综合征(KSS)。③mtDNA拷贝数突变,拷贝数大大低于正常,较少见,见于致死性婴儿呼吸障碍、乳酸中毒等。
4.儿科常见的几种线粒体病
线粒体呼吸链由5种酶复合物组成,它们由mtDNA和核基因共同编码。所以,编码这些酶的核基因突变也能产生线粒体病的表型,使线粒体病的遗传学病因诊断困难。线粒体病种类多,原因各不相同。下面列出几种儿科常见的线粒体病:
(1)Leigh综合征(Leigh syndrome)
又称亚急性坏死性脑脊髓病,是一种高度遗传异质性疾病。线粒体呼吸链中5种酶复合物中任意一种酶的功能障碍均可导致该病发生,其中约20%由mtDNA突变所致,常见突变A3243G、A8344G、T8993G/C。临床主要表现:共济失调、肌张力低或肌张力异常、肌强直、发育迟缓或倒退、视神经减退、眼外肌麻痹、发病年龄早,患儿多早期死亡。MRI基底节和脑干长T1、长T2信号。实验室检查对本病诊断有重要意义,血、尿、脑脊液的乳酸、丙酮酸的浓度明显增高,丙酮酸更显著;代谢性酸中毒;血和脑脊液氨基酸分析示丙氨酸浓度增高。丙酮酸脱氢酶和线粒体呼吸链复合物酶活性测定对判断突变基因有帮助。治疗:辅助对症,营养支持,预后差。
(2)线粒体脑病-乳酸酸中毒-卒中样发作综合征(mitochondrial encephalomyopathy with lactic acidosis and stroke like episodes,MELAS)
多由mtDNA突变所致,表现为母系遗传,常见突变A3243G占约80%。临床主要表现:发病年龄3~13岁,发病前通常无明显发育迟缓,有矮小和多毛,就诊常因出现中枢神经系统症状,如脑卒中样发作的偏瘫、构音障碍、脑病、偏头痛样发作、视力障碍等,可反复发作,常有惊厥发作;累及多系统和器官,表现复杂。实验室检查:血、脑脊液的乳酸、丙酮酸的浓度增高,有间断性发作性特点,血氨增高。肌活检:RRF(+)。MRI:卒中样发作时枕区长T1和长T2,MRA无血管狭窄和闭塞;随病程基底节长T1和长T2,脑萎缩。治疗:无特效方法,二氯乙酸可减低乳酸和部分改善症状。抗癫痫治疗。预后差,发病数年内死亡。
(3)肌阵挛性癫痫伴破碎红纤维综合征(myoclonus epilepsy and ragged red fibers,MERRF)
多由mtDNA突变所致,表现为母系遗传,常见突变A8344G占约80%,该突变阈值高,大于80%影响线粒体功能。临床主要表现:儿童期发病,肌阵挛癫痫、共小脑济失调、智能减退,听力、视力下降,周围神经病。实验室检查:血乳酸、丙酮酸浓度增高,脑脊液可正常;肌活检:RRF(+);MRI:大小脑萎缩。治疗:无特效方法,抗癫痫,辅酶Q、叶酸、肉碱、维生素辅助治疗,儿童预后差。
(4)线粒体缺失综合征(mitochondrial DNA deletion syndrome)
包括三个临床相关的综合征,Kearns-Sayre综合征(Kearns-Sayre syndrome,KSS)、进行性眼外肌麻痹(progressive external ophthalmoplegia,PEO)和Pearson综合征,三个综合征的表型可出现在同一个家庭的患者中或同一个患者的不同病程中。由mtDNA缺失突变所致,缺失大小在2~10kb。最常见的有两种缺失:其一8482~13 459间缺失4977bp,缺失两端有 13bp的同向重复序列;其二10 204~13 761和10 208~13 765间缺失35 57bp。多为新生突变。KSS主要表现视网膜色素变性,眼外肌麻痹、上眼睑下垂、复视、心脏传导阻滞,其他智力减退,小脑共济失调,听力下降,内分泌系统可见胰岛素分泌不足引起的糖尿病,甲旁腺功能低下等。PEO发病较迟,主要眼部症状,重症发展成 KSS。Pearson综合征发病早,2岁内,表现铁粒幼细胞贫血和胰腺外分泌功能异常,存活至10岁后出现KSS。实验室检查:血氨升高,血、脑脊液的乳酸、丙酮酸的浓度增高;肌活检:RRF(+);骨髓:Pearson综合征铁粒幼细胞贫血和铁染色线粒体中异常沉积;MRI:脑干、小脑萎缩,基底节改变。治疗:无特效方法。对症,辅酶Q、叶酸、维生素辅助治疗,胰酶辅助治疗消化不良,必要时安装心脏起搏器。预后不良。
(四)非经典孟德尔遗传
大多数遗传病特别是单基因病遵循孟德尔遗传规律,但是有一些遗传病有其他的遗传规律,包括前面讲到的动态突变、基因印迹、单亲二体和表观遗传机制。
(五)X染色体失活
1.Lyon假说
近半个世纪前,英国遗传学家Mary Lyon提出X染色体失活(X-inactivation)的假说,即Lyon假说。要点有:雌性哺乳动物体细胞的两条X染色体中有一条没有遗传活性;失活的染色体是随机的,既可来自父亲也可来自母亲;失活发生在胚胎早期,失活一旦发生,由该细胞繁衍的子细胞都具有同一条失活的X染色体。女性一条失活的X染色体,在细胞间期异固缩,位于核膜内缘,为深染的染色质,被称为Barr小体;X染色体失活发生在胚胎早期,始于桑葚胚期,约在合子后64~100细胞阶段(孕16天)完成。女性进入性成熟期后,处于失活状态的X染色体在随初级卵母细胞进入减数分裂时恢复生物活性。故在桑葚胚期之前女性的两条染色体都具有活性。女性体细胞X染色体失活后,在以后的细胞有丝分裂过程中不再复活。X染色体失活的过程称之为Lyon化(lyonization)。
2.X染色体失活的调控机制
X染色体失活由表观遗传学控制,目前尚未完全清楚。X染色体失活受位于Xq13的失活中心(X-inactivation center,XIC)和所包含的XIST(X-inactive specific transcript)基因调控。XIST编码17kb非翻译RNA,由失活的X染色体表达,目前研究说明其作用主要是启动静止过程。在XIC还有调节元件,如TSIX及相关因子,TSIX也是一个非编码的RNA,为XIST的反义抑制子。此外,组蛋白的修饰、CpG甲基化、与染色质结构相关的蛋白质的作用等均参与X染色体失活过程。
失活X染色体的基因大部分失去活性,不具有转录功能。人约15%的X-连锁基因逃避失活,仍然保留转录活性(为X染色体的双等位基因表达),但是失活的X染色体的基因表达水平很少能达到有活性的X染色体表达的量。近来,用基因芯片技术鉴定:女性相对于男性只有少量的X-连锁基因过度表达,也说明完全逃避失活的基因很少。逃避失活的基因在X染色体不是随机分布,大部分逃避失活的基因成串地位于X染色体的短臂,多与Y染色体有同源性。
3.X染色体失活的遗传学意义
①基因剂量补偿作用:女性有两条X染色体,而男性只有一条X染色体,由于女性两条之一是没有活性的,使男女X连锁的基因表达趋于平衡。②嵌合体(mosaic):女性两条X染色体随机失活,即失活的X染色体既可能来源于父亲也可能来源于母亲,使女性体内部分细胞带有母源的X染色体有活性,而另外一些细胞带有父源的X染色体有活性,成为同一个体带有两种不同亲代来源X染色体的嵌合体,表型取决于体内两种细胞群的比例。③与Lyon假说不相符的遗传现象:X染色体失活偏斜(skewed X chromosome inactivation)。在X染色体发生结构畸变,如缺失或重复时,该X染色体常常处于失活状态;X染色体与常染色体发生易位时产生的衍生染色体往往保持有活性,而没有发生易位的X染色体处于失活状态。一些X-连锁的单基因病,如Rett综合征、X-连锁隐性遗传病DMD的女性携带者患病均认为与X染色体失活偏斜有关。因此,对累及X染色体的遗传病,在细胞或分子水平分析X染色体失活状态是否发生了偏斜,对临床分析表型特点均可提供帮助。
近年来,对遗传与先天代谢性疾病的治疗方面有了较大的进展。治疗的方式包括“环境工程”和“基因工程”两大类,前者系通过改善内、外环境因素如通过饮食、药物、手术、脏器移植等以纠正代谢紊乱,改善症状;后者系用人工方法改造和修补有缺陷的基因,以期达到治疗的目的。
(一)饮食及药物疗法
原则是补充缺乏物质,避免和除去有害物质。
1.补充缺乏物质(补其所缺)
补充人体代谢所需要的物质如维生素、电解质、氨基酸、葡萄糖等。另外,根据遗传病的病因给患儿针对性地补充所需成分。详见各章节相关疾病的治疗。
2.避免摄入有害物质(禁其所忌)
包括避免摄入或摄入后在体内代谢产生过多的无用或有害产物的物质,如半乳糖血症应在新生儿期即禁食含半乳糖、乳糖的人乳或牛乳,代以谷类食品;苯丙酮尿症采用低苯丙氨酸饮食。目前针对不同的代谢病设计了100多种奶粉和食谱,开始治疗的时间越早,患儿年龄越小,治疗效果越好。肝豆状核变性应限制铜的摄入;G-6-PD应避免食用蚕豆、菠萝、氨基比林、抗疟药;Lesch-Nyhan综合征避免嘌呤食物等。
3.排除过多有害物质(去其所余)
采用物理化学的方法将过多的毒物排除或抑制生成。用促排泄剂、螯合剂、代谢抑制剂、平衡清除法、换血或血浆置换过滤等方法减少体内的毒物,以缓解症状。如高胆固醇血症用胆固醇胺螯合剂以抑制胆红素的肠肝循环,从而减少脂肪吸收。口服D-青霉胺与铜离子结合,使肝豆状核变性消除过多铜的沉积,减轻症状。高胆固醇血症、溶酶体病如脂质沉积病、黏多糖病、黏脂病等,以及某些溶血性贫血可以输血浆、进行血浆置换,以减少血中过多蓄积物质;亦可采用血浆过滤,并可再输入含有肝素、肝素琼脂糖小球和氯化钙的患者血浆,使血浆中的低密度脂蛋白结成不溶复合物不再进入患者体内,使高胆固醇血症得以降低。使用代谢抑制剂,如应用别嘌呤醇抑制黄嘌呤氧化酶治疗Lesch-Lyhan综合征减少尿酸生成,缓解症状。
(二)酶疗法
供给必需的酶,纠正代谢缺陷。
1.酶诱导
利用药物或其他方法增强体内酶的活性或增加其合成,如用苯巴比妥可使肝中葡萄糖醛酰转移酶活性增强,治疗Crigler-Najjar综合征、Gilbert综合征等新生儿黄疸。
2.酶补充
补充缺乏的酶,如黏多糖贮积症Ⅰ型使用loronidase替代治疗对非脑部症状有效;黏多糖贮积症Ⅱ型(Hunter)无神经系统症状者使用idursulfase替代治疗;Fabty综合征用agalsidaseα/β替代治疗均证明有一定疗效。
(三)外科疗法
1.脏器移植
从种意义上讲,移植脏器是单基因病的终极遗传学的治疗,可代替脏器的作用,产生酶和基因产物如激素、免疫物质、凝血因子等以维持正常代谢,或将蓄积物质分解排泄而起到治疗作用。临床应用肾、肝、脾、肺、心、胸腺等器官移植取代患病器官。但器官来源是非常困难和有限的。如何解决器官来源是一个重大的课题。
2.干细胞移植
干细胞是非特异化的细胞,具有自我再生和分化成特异细胞系的潜能。有两大类:体干细胞和胚胎干细胞。体干细胞分造血干细胞和非造血来源的体干细胞。造血干细胞用于临床40多年,能分化成粒系和淋巴系的造血干细胞,可用于一些早期的溶酶体贮积症和脑白质营养不良患儿的治疗。非造血来源的体干细胞来源于各脏器官,尽管体干细胞可以分化为其来源器官的多种细胞系,是否可以分化成其他器官的细胞还不肯定。胚胎干细胞最早来源于对小鼠畸胎瘤的研究,发现其中存在未分化的多能干细胞,这些多能干细胞在一定的条件下能分化成为多种类型的细胞。人胚胎干细胞(human embryonic stem cell,hES)是一种源于人囊胚内细胞团,经体外分离、培养获得的原始多能干细胞。由于其人源性,人胚胎干细胞受到高度关注,潜在用途是通过定向分化诱导产生各种特异的细胞和组织,将其用来修复或替换丧失功能的组织和器官,目前这部分内容仍在研究阶段。
3.矫形手术
通过手术的方法矫正各种畸形,帮助恢复机能。
(四)基因工程与基因治疗
遗传工程包括细胞工程、染色体工程、细胞器工程和基因工程,就是采用类似工程技术的方法,人工合成或分离出全部、部分或单个基因,在体外进行重新组合,然后转移到人体或生物体内,定向改造生物的遗传特性。也就是人工造成遗传信息的改变,使病变的单个或多个基因得以修复或被代替或关闭抑制获得正常功能或转移全部基因,克隆出新的生命,这类技术统属于基因转移技术(gene transfer technique)。采用基因转移技术治疗疾病称为基因治疗(gene therapy)。有可能进行基因治疗的遗传病必须符合以下条件:①疾病发病的分子机制及致病基因的分子结构清楚;②目的基因能被克隆、其表达调控机制已较清楚;③该基因可在体外和体内进行繁殖和表达;④有安全可靠的载体包装细胞系;⑤基因转移细胞有极好的繁殖力并在体内能长期存活;⑥目的基因在受体细胞内能完整、稳定、剂量适当地整合和表达。
基因治疗基于遗传病的病理基础是基因突变,纠正这种异常即可使疾病获得根治的理念。由于遗传病机制不同所采用的策略不同。基本策略有:①基因修复:修复缺陷基因使其在质和量均能够正常表达,目前尚无法做到;②基因替代:去除整个异常基因,以功能正常的基因代替,使致病基因得到永久更正,目前也无法实现;③基因抑制或失活,导入外源基因,抑制异常基因表达;④基因增强:将目的基因导入异常细胞或其他细胞,使目的基因的表达产物可以补偿缺陷细胞的功能或使原有功能得到加强,适于单基因隐性遗传病的治疗,目前基因治疗多为此类;⑤重新开放已关闭的基因:目的在于促使有类似功能的基因表达已超过或代替异常基因的表达。如去甲基化使关闭的γ珠蛋白基因重新开放,合成HbF(α2γ2)治疗β地中海贫血。
基因治疗有两个目标:其一,生殖细胞的基因治疗,是将正常基因转移到突变的生殖细胞(精、卵细胞或受精卵)使其发育成为正常个体,并且可使有害基因不再传递给下一代,这是最理想的根治遗传病的方法,但目前还不能实现。其二,体细胞的基因治疗,是将目的基因转移到体细胞使其发挥作用来达到使患者症状消失或减轻的治疗目的,突变的致病基因仍然可以传递给下一代。体细胞基因治疗最理想的状态是将正常基因导入靶细胞内染色体特定的基因座实现正确表达,但目前染色体定位转移仍有很大困难。实现这两个目标都需要将正常的基因转移到受体细胞,并使其在受体细胞正确地表达。所以外源基因的安全导入和高效的正确表达是基因治疗的关键。基因治疗的方法:
1.基因转移
有基因体外输入和体内输入两种方式。体外输入限于可从患者体内取出,经过体外基因修饰后回输入体内的细胞群,如成纤维细胞、肝细胞、内皮细胞、肌细胞和骨髓干细胞等。回输后有选择优势的细胞更适合,如利用骨髓干细胞治疗联合免疫缺陷症;治疗血友病的Ⅷ因子自体成纤维细胞回输。体内输入是直接基因输入,理论上可以治疗很多遗传病。但靶向基因应该满足能高效转移到个体细胞并整合到基因组,长期、稳定表达的条件,目前还很难达到全部要求。因此,探索理想基因转移方法仍是基因治疗的一项重要内容。方法有
(1)物理法:①显微注射:显微镜下向细胞核内直接注入外源基因,一次只能注射一个细胞,工作耗时费力;②电穿孔:利用脉冲电场提高细胞膜的通透性,使细胞膜上形成纳米大小的微孔,将外源DNA转移到细胞中,但有时会损伤细胞;③微粒子轰击:利用微小的金、钨等贵金属颗粒吸附DNA,在高压作用下将DNA伴金属颗粒高速导入细胞,可以使DNA在活体组织、贴壁细胞和悬浮细胞中表达;④脂质体法:用人工的脂质体包围外源基因,再与细胞融合,或直接注入病灶组织使其表达。
(2)化学法:将外源基因与带电荷的物质磷酸钙、DEAE葡聚糖、聚L鸟氨酸和聚卤化季铵盐或若干脂类混合,形成沉淀的DNA微颗粒,加强细胞摄取外源DNA能力,但其转移效率低。
(3)生物学方法,用病毒载体介导基因转移,有RNA病毒和DNA病毒。病毒载体是目前最有效转移目的基因的方法,最常用的病毒载体是反转录病毒和腺病毒。①反转录病毒(restrovirus,RV)载体:RV是RNA病毒,有反转录酶,使RNA转录为DNA,再整合到宿主细胞基因组,其具有转染效率高、稳定表达外源基因和有广泛宿主细胞的优点,但有容量小和可使细胞恶变的缺点。最常用的病毒是莫洛尼鼠白血病病毒(murine leukemia virus,MO-MLO)。近年慢病毒(lentivirus)也受到关注,有可以感染不分裂的细胞、效价高、主要的目的基因表达时间长的优点。②腺病毒(adenovirus,AD)载体:AD是DNA病毒,无细胞膜,多数AD载体以2型和5型为AD基础构建,由于缺失E1和E3,自身不能表达包装蛋白,需要293细胞或Hela细胞的帮助形成病毒颗粒从而感染细胞。插入DNA片段长,不需要分裂的靶细胞,有原位感染的能力和病毒滴度高的优点,一般不整合到宿主的基因组,减少了插入突变的潜在危险。已用于肺、肝、中枢神经系统、内皮细胞、肌细胞和唾液腺上皮细胞的基因治疗实验,对呼吸系统可以反复多次滴注,临床用于囊性纤维化的治疗。
(4)同源重组:哺乳类与细菌或酵母菌一样有酶系统,可使进入细胞的外源基因和染色体上的基因在同源序列间发生重组而插入染色体,外源基因就可以专一地整合到细胞的特定位点,从而避免了因随机插入得不到调控和表达,甚至可能激活或失活插入位点附近的基因等麻烦问题。基因修正主要利用这种方法。目前由于体细胞不允许在体外长时间培养、同源重组率低和筛选工作量大的缺点限制了临床使用,但因其能够定点插入目的基因,避免了很多的副作用,随着技术方法改进,增加同源重组率,此方法仍有较大的应用前景。
总的来讲,人类实验表明输入基因并使其持续表达以诱导表型改变至少在基因表达水平可行,最大的挑战是如何使输入的基因表达水平达到使表型改变的水平?如何不诱发宿主的防御系统反应?如何减少整合性载体的插入诱变?以及如何使被纠正的细胞有后续的选择优势或持续增殖?逐步解决了这些问题基因治疗才能更多地应用于临床。
2.RNA修饰治疗
这类方法着眼于mRNA,抑制mRNA表达或改变或增加mRNA功能的方法。
(1)反义寡核苷酸(antisense oligonucleotide,ASO)
ASO是单链DNA,含18~30bp,通过碱基互补原则与目标mRNA结合,降低基因表达水平。理论上过量表达突变蛋白所致疾病均适用,但目前其在细胞的稳定性、摄取率、结合靶序列的特异性和亲和力方面都有有待解决的问题。如已开展的DMD临床前试验,在前mRNA剪切时诱导外显子跳过(exon skipping),从而恢复读码框并产生一个截短了但仍有部分功能的蛋白。
(2)RNA干扰(RNAi)
RNAi是自然存在的一个形式,可以介导mRNA降解或翻译阻滞。主要挑战是如何提高有效性、特异性和准确地导入靶细胞。
(3)反式剪接(trans-splicing)
调控前 mRNA (pre-mRNA)水平,目标基因的前mRNA被反式剪接改变成另一个独立的前mRNA,然后用载体将其输入体内。节段性反式剪接(segmental trans-splicing,STS)解决了对传统病毒载体来讲太大的基因转入,如Duchenne肌营养不良基因,STS包括传输2个独立的基因转移载体,每一个编码独立的前mRNA,然后通过剪接体介导的反式剪接产生完整的前mRNA。
(4)核糖核酸酶(ribonuclease)
它是有酶活性的RNA分子,识别特异性RNA序列,并在目标分子内进行位点特异性的磷酸二酯键切割。结构包含两个反义RNA区域(二翼互配区)包围在核苷酸水解基序(nucleotide hydrolysismotif)的两侧,提供作用的靶特异性。
对于RNA修饰治疗的临床应用最大障碍是转移问题,如何提高转移效率使其达到治疗作用将有赖于基因转移载体的发展与进步或其他突破性的研究工作。其过程如图30所示。
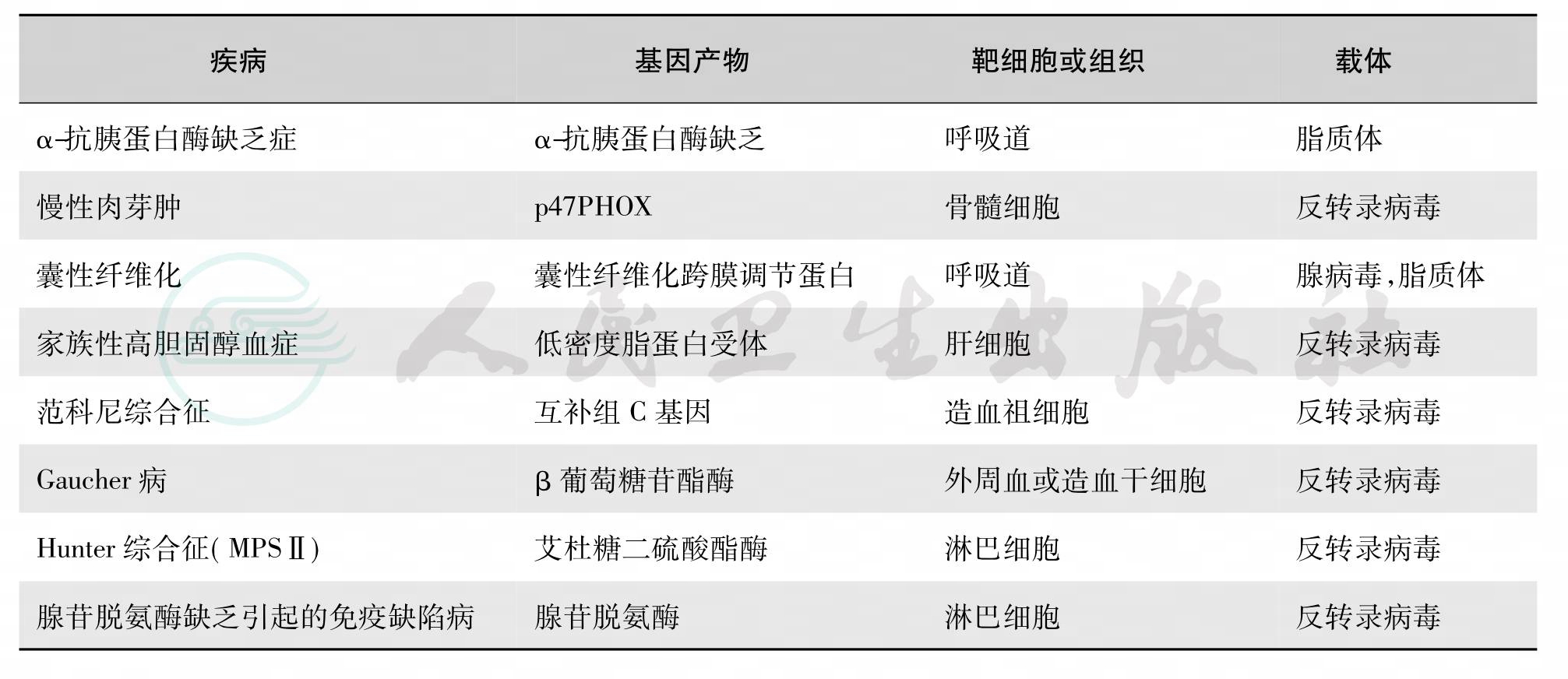
基因治疗的临床应用目前还存在很多的问题,主要包括稳定性、安全性、转移基因的高效表达问题、免疫性和伦理等需要研究解决。目前发现的遗传病有近6000种,由于各种原因的限制只有20多种被列为基因治疗的主要对象,部分进入临床试验阶段。尽管如此,它作为遗传病治疗的新手段正在逐步被人们所接受,它蕴藏着巨大的潜力,随时间将证明遗传病是可以治疗的(表11)。
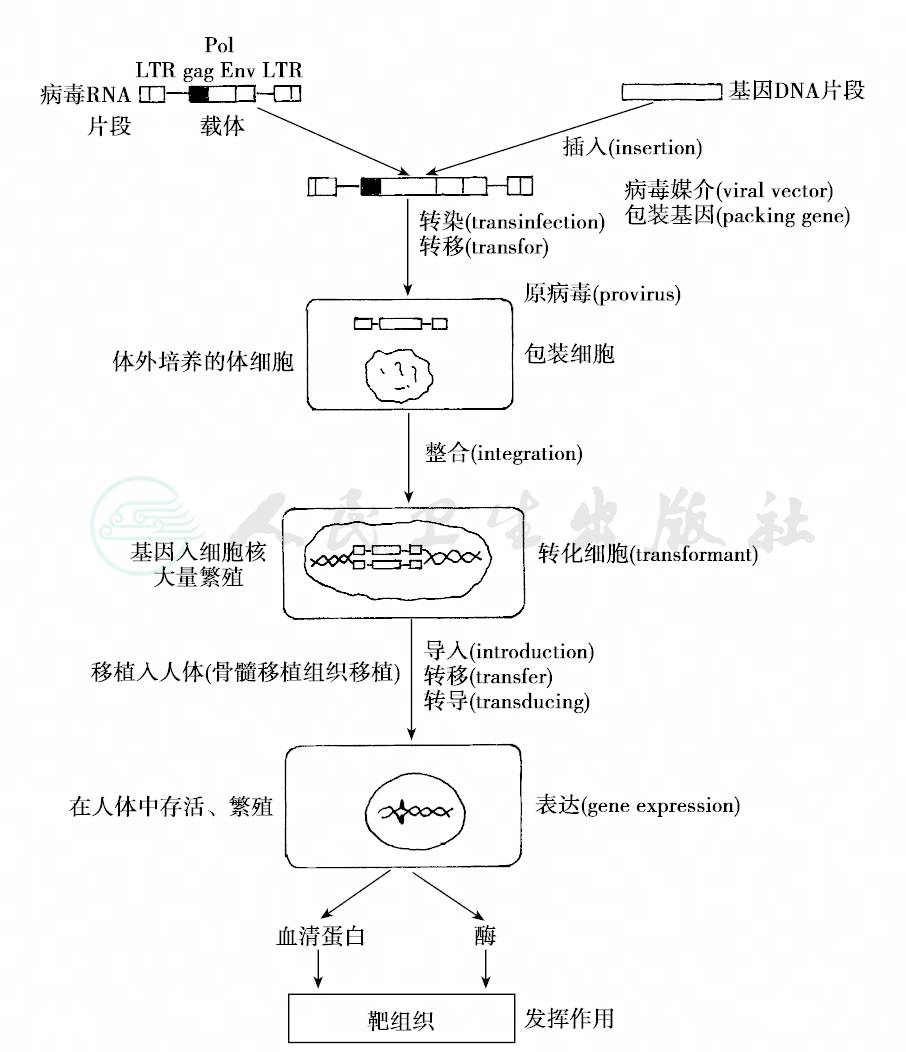
图30 基因治疗过程图解
LTR侧翼顺序(含启动子、增强子及终止子),gag衣壳,Pol逆转录酶,Env包被,以上俱为载体病毒的结构
表11 目前临床试用的基因治疗的遗传病
优生科学(birth health science)是一门综合性科学,它来源于1883年英国Galton提出的优生学运动,但其被法西斯和统治者所利用,制造了种族灭绝和专制压迫剥削的借口和恶果。“eugenics”一词已为人们所摒弃。我国的优生科学则是继承了我国古代和历代优生思想,汲取国外的分子医学、生殖健康、围生医学等近代知识,以生物学、医学、环境学和遗传学为基础,在社会、经济、环境、文化和伦理的优良条件下,采取遗传咨询、产前诊断、植入前诊断、预防不健康的有遗传病和先天性缺陷与残疾儿的出生,并积极关注孕期、围生期、新生儿期的保健以及婴幼儿期、青少年期的健康和教育,达到提高人口素质(improving human quality)的目的。
从遗传学的观点看,我国各民族既有大量的优质基因,也有一定数量的有害基因,两者构成我们的基因库,有害的基因即遗传或基因负荷。采取有效的优生措施,使优秀的基因得以保存传递,使有害的基因趋向减少和有效控制。重视遗传医学,防止和减少有严重的遗传性或先天性疾病的胎儿出生是一项重要的工作。但防止遗传不良因素尚不是唯一内容,因为怀孕后孕妇营养不良、感染、滥用药物、环境污染、接触有害物质以及母亲患有各种疾病等,都可以影响胎儿在宫内的生长和发育。分娩时难产引起的窒息和颅内出血,以及其他产伤等也可以影响新生儿的成长和智能发展。新生儿出生后护理不好、营养不良发生各种疾病也难以使新生儿成长和发育正常。为此,要做好优生工作,除了要研究和实施遗传医学外,还要做好围生医学和新生儿保健工作。通过遗传医学防止和使早期发现有遗传病和有严重先天性缺陷的胎儿或患儿,经过选择性出生或早期干预治疗;通过围生医学做好孕产期保健;通过新生儿保健做好新生儿的护理,这些都是重要的环节。所以,我国推行的优生学是一门广义的优生科学。优生科学的措施,可分遗传医学、围生医学和生长发育期措施几个方面。
(一)遗传医学方面的措施
1.全面遗传学普查 研究优生科学应从各地区的实际需要出发,从调查劣生着手,组织力量,有计划、有步骤地在国内进行各省市遗传病的普查,摸清遗传病种类、分布和频率。特别着重在严重遗传病和其携带者的普查,了解患病率和突变的携带频率,找到切实可靠的科学依据,以便设计有效的防治措施。例如,已经熟知广东、广西和海南地区存在着较多的地中海贫血和血红蛋白病,需要实行产前筛查和诊断;成都市0~6岁儿童出生缺陷和遗传学特征调查协作组,对11 230儿童进行全身各系统的先天畸形或发育异常的调查,以后又推广到全国29个省(直辖市、自治区),特别注意到较多发的神经管缺陷调查、缺碘性甲状腺肿等多发性疾病,也获得了不少资料,为优生、优育提供了有益的根据和建议。近年来,通过给育龄妇女免费发放叶酸,同时加强知识普及和孕妇教育使神经管缺陷的发生率明显减少。
2.禁止近亲结婚 对已经查明的遗传病患者或携带者,应禁止直系亲属内和三代以内的旁系亲属内结婚。若父或母是显性遗传病患者,则代代均有50%的患病风险,若父母均是隐性遗传病患者的亲属,父母双方各带一个致病隐性基因的可能性很大,结婚后所生子女的患病风险为25%。死胎和婴儿病死率也比正常高出1.5~2倍。贵州省一些民族的近亲结婚情况调查也说明了这点。应该指出,无血缘关系的人之间婚配在极大程度上能使携带者的子女少发生遗传病,但不能减少人群中这个有害基因的频率,该基因仍继续存在和传递。此外,按婚姻法规定,禁止未经治愈的麻风病、精神分裂症患者或者其他在医学上认为不应该结婚的疾病患者结婚也是很必要的。
3.为了防止遗传病患者在下一代再现以及保证胎儿和儿童健康发育成长,可采取下列措施:
(1)婚前检查:在我国婚前体检是一种自愿行为,但作为把握人口素质的第一关,婚检又是十分必要的。在一些地区提供免费服务,内容包括体格检查、健康询问及调查家族史等。医务人员应根据各种有碍优生的疾病,包括遗传病进行相关的宣教,对有遗传病或有遗传病家族史者建议他们进行遗传咨询。
(2)适龄生育:染色体病,如最常见的21-三体综合征的患病率与母亲年龄密切相关,母亲20~30岁时患病率1/800~1/1000,35岁以上时为1/350,40岁以上时为1/100,45岁时为1/20。这是由于卵子老化,引起染色体不分离的结果。因此,母亲生育第一胎的年龄不要超过35岁。
(3)设立遗传咨询门诊:对遗传病基因携带者和可能生育遗传缺陷儿的父母及有遗传病家族史的青年对象进行遗传检测和咨询。其工作内容除了一部分属于询问高龄婚配、近亲婚配、反复性流产,以及服用药物或接受放射性剂量后有否潜在危险等的询问外,主要的工作重点是对遗传病做出诊断或明确诊断,提出优生建议。
遗传咨询是医务工作者的职责之一,特别是儿科医生应具备遗传学知识,当遇到下列情况:①患遗传病或家庭成员患有遗传病;②家族中多成员多次出现原因不明、表型相似的疾病;③有发育异常的孩子;④疑有染色体异常的家庭或母亲有非妇科原因的反复流产;⑤性发育异常或不孕症以及近亲结婚,应进行家系分析并指导遗传就诊。对于一些遗传规律较明确的遗传病,根据疾病特点提供较确切的风险评估。
(4)一般无临床表现而可能为突变基因携带者,主要是检出隐性杂合子。检出方法如前所述有:①突变基因检测;②直接测定酶活性,可能减低;③测定生化改变,贮积物的量常介于正常人与患者之间;④观察某些不令人注意的病态特征,根据家庭出现的表型做出判断。
隐性遗传病中有些杂合子携带者可以做出诊断,如血友病A、B携带者的血中Ⅷ及Ⅸ因子减少、苯丙酮尿症携带者的血中苯丙氨酸含量高于正常、Duchenne肌营养不良携带者血磷酸肌酸激酶增高、葡萄糖-6-磷酸脱氢酶缺乏症携带者高铁还原试验阳性、抗维生素D佝偻病携带者血中磷酸盐减少、丙种球蛋白缺乏症携带者血中丙种球蛋白缺乏、纤维蛋白原缺乏症携带者血中纤维蛋白缺乏、半乳糖血症携带者的尿中还原物质增加、肝豆状核变性携带者血铜蓝蛋白减低或尿铜增加等。对于常染色体隐性遗传病,若父母同是杂合子,其下一代将有25%的患病风险;X-连锁隐性遗传病,母亲若是杂合子,其子将有50%的患病风险。因此,如能在孕前检出杂合子对这个家庭非常重要。例如,苯丙酮尿症是一种严重的氨基酸代谢异常的遗传病,可使患儿智能发育障碍。携带者血浆中苯丙氨酸含量升高,与正常人有一定程度的重叠,给可疑携带者口服一定量的苯丙氨酸后测定血浆中苯丙氨酸在一定的时间内的降低速率,杂合子耐量曲线在正常人与患者之间。因此,对一些先天性代谢缺陷,检出携带者也是有效措施,可以防止患儿的出生。
(5)产前检查:孕妇自怀孕后就可以进行产前的检查。针对不同孕妇群体产检项目提供不同层次的服务。适龄孕妇应进行的常规孕检及B超检查和唐氏筛查等。B超检查可以对整个孕期,即从胚胎至胎儿期监测胎儿生长发育情况,有3个重要的时间窗口:早孕期11~12周,检测胎儿颈部透明层厚度和鼻骨发育(与21-三体胎儿及其他染色体病相关),发现胎儿严重畸形,如无脑儿等;孕中期22~24周,发现大部分胎儿的结构畸形;孕晚期32~36周,评价胎儿发育情况和在孕晚期出现或才能诊断的胎儿畸形。唐氏筛查可以提示出21-、18-三体和开放性神经管缺陷及其他一些染色体病的高风险胎儿,有早孕期筛查、早孕联合中孕筛查、中孕筛查。
(6)产前诊断:有相应的适应证和方法。近几年非侵入性产前诊断随分子生物学技术的快速进步有了很大的发展,如用从孕妇外周血中分离出的胎儿细胞进行单细胞全基因组的SNPs和CNVs分析;用孕妇外周血中的胎儿游离DNA(cell free fetal DNA,cffDNA)检测胎儿染色体异常,对21-、18-和13-三体检出率达到98%以上,是目前最有效的产前筛查方法。随着技术的进一步提高,有望拓展到DNA片段性缺失、重复等染色体微小结构重排和单基因突变的检测。
(7)孕期保健:包括早期诊断妊娠,定期产前检查,治疗各种孕妇并发症,防止对各种诱变剂的接触,如X线、中子、质子等辐射;秋水仙碱、亚硝胺、氮芥,某些抗生素,农药,食物防腐剂、着色剂,咖啡因和工业毒物等。防止病毒感染:甲型肝炎、流感、麻疹、风疹、单纯疱疹等,均可能引起胎儿畸形,特别是在孕早期危害更大。禁止酗酒、吸烟和吸食毒品,注意孕期营养和精神卫生,及时识别高危孕妇和发现胎儿生长迟缓,防止难产。另一方面要普及胎盘-胎儿功能和胎儿成熟度测定,防止早产、过熟儿和产伤儿出生。重视指导胎教,使孩子活泼、健康、聪明。
(二)围生医学方面的措施
1.分娩监护
重视观察产程,注意胎心和胎动,早期发现胎儿宫内窘迫,防止难产所造成的产伤和新生儿窒息,以减少伤残和智能方面的不良影响。
2.新生儿保健
包括新生儿窒息时进行复苏,早产儿的护理,畸形儿的早期发现和处理,保证新生儿的营养,提倡母乳喂养,重视观测新生儿行为如醒觉、睡眠、摄食、啼哭、活动、视、听、爱抚、刺激等的反应、检查身体和精神状况,各种生理反射,有无病理反射。按计划免疫进行免疫接种。注意保暖、防暑、提倡早期活动等。
(三)生长发育期的措施
由婴儿至青春期进行,提倡改善人类的环境来发挥人类的智能及生物潜力,使人类的身心健康达到更完美的境地(improving human quality)。预防治疗或改变环境条件以控制表型的形成,达到补偿或挽救某些遗传缺陷的个体。例如对苯丙酮尿症患儿早期筛查、施行饮食控制疗法,使患儿智能不受影响,达到正常人的水平。对呆小病患者早期应用甲状腺素,地方性甲状腺肿给予碘,脱离或防止接触毒物、污染、服用有害食物、药物,避免吸烟、酗酒等不良习惯的养成,也能达到同样效果。有不少人主张,在胚胎期及出生后的半年至一年间,加强营养,可使脑细胞增殖得更多一些,从而在不改变基因型的情况下,使婴儿的智能发育更臻完美。对于一些先天畸形可以进行手术矫正。年龄较长则是主张通过教育和心理学辅导各方面的措施,包括家庭、学校、社会的共同教育和影响、优生政策、法律的实施,达到家庭和谐、邻里相助、学校关切,使其德、智、体、美得到全面发展、生物潜能得到充分发挥,人口素质日益提高。以上都不仅涉及人体遗传组成,而且是通过环境影响以进一步改善遗传素质,皆属于优生的范围,也是优生科学的发展方向。









