


 收藏
收藏 已收藏
已收藏英文名称 :spinalmuscular atrophy
脊髓性肌萎缩(spinal muscular atrophy,SMA)是运动神经元存活基因1(survival motor neuron,SMN1)突变所导致的常染色体隐性遗传病,SMN1基因突变导致脊髓和脑干运动神经元变性,临床上主要表现为近端肢体和躯干进行性、对称性肌无力和肌萎缩,智力认知正常,感觉无受累。随着疾病的进展,可累及呼吸系统,重症患儿常死于呼吸衰竭。SMA在活产婴儿中的发病率约为1/6 000~1/10 000,中国人群中的携带者频率约为1/42。临床表现为一个连续广泛的疾病谱,一般根据起病年龄、肌无力严重程度和所获得的最大运动功能,将SMA由重到轻分为五种不同的临床类型,即SMA 0~4型,其中以SMA 1型(婴儿型)最常见。
1990年 Brzustowicz及 Melki等首先将SMA致病基因定位于5q11.2~13。1995年初Lefebrre等分离克隆出SMA致病基因,命名为 SMN。SMN1基因有9个外显子,大约有95%的SMA患者为SMN1基因第7或第7、8外显子纯合缺失突变,5%为复合杂合突变,由此导致SMN蛋白表达量下降。SMN蛋白广泛存在于神经元细胞质、细胞核和神经轴突中,并参与核糖核蛋白的传递及相互作用,对运动神经元的功能至关重要。SMN蛋白的缺失导致了肌动蛋白的减少和运动神经元的生长障碍,从而导致脊髓性肌萎缩的发生。
SMA的修饰基因SMN2与SMN1基因高度同源,SMN1基因位于端粒侧,修饰基因SMN2位于着丝粒侧,两者间有5个碱基的差异,约10%左右的SMN2 premRNA可被正确剪切而表达全长有功能的SMN蛋白,在SMN1基因缺失时起剂量补偿作用。故SMN2基因拷贝数与SMA表型严重程度呈负相关:SMN2基因拷贝数在1型患儿中多为2拷贝,在2型患儿中多为3拷贝,在3型患儿中多为3~4拷贝,4型患儿中多>4拷贝,但SMN2拷贝数并不能完全预测表型严重程度。
脊髓病理检查可见脊髓前角细胞变性和减少。肌肉活检光镜检查可见肌纤维大小不等,肥大与萎缩肌纤维混杂分布,呈特征性的群组化分布特点(见图1)。SMA 1型肌纤维萎缩更严重,群状或束状萎缩明显。可见少许炎性或变性坏死性细胞。电镜可见脊髓的神经元减少,神经纤维稀疏,髓鞘崩解,轴索萎缩,间质组织增多。
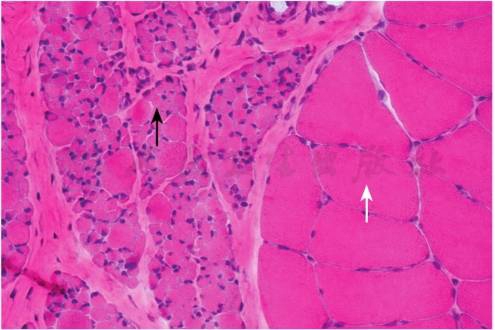
图1 脊髓性肌萎缩患儿肌肉活组织检查
HE染色光镜观察(400×),肌肉细胞呈典型的群组化特征,可见异常肥大的肌肉细胞(白箭头)和萎缩的肌肉细胞(黑箭头)。肌肉细胞间的结缔组织增多。
血清肌酸激酶一般正常或轻度升高。肌电图自发电活动(正相、束颤、纤颤电位)明显增多。轻收缩时,运动单位的电位时限延长,波幅增高,典型者可见巨大的动作电位,重收缩时运动单位数量减少。运动神经传导速度正常或轻度减慢,感觉神经传导速度检查正常。基因检测是目前主要的确诊手段,常采用多重连接探针扩增法(MLPA)、实时荧光定量PCR(qPCR)、PCR限制性酶切分析法(RFLP)和变性高效液相色谱(DHPLC)等检测SMN1基因第7或第7、8外显子缺失突变。SMN1基因测序用于检测SMN1基因内是否存在微小突变。肌肉活组织检查仅用于少数基因检查正常的患儿,观察有无典型的脊髓前角病变,从病理学上确诊。
既往本病除对症支持治疗外,无特效治疗方法。随着分子修饰治疗和基因治疗的进展,通过调节SMN2基因表达和SMN基因替代治疗以提升SMN蛋白表达,已成为临床上治疗SMA的主要方向。反义寡核苷酸药物诺西那生通过改变SMN2基因剪切,增加全功能性SMN蛋白,明显改善患者的运动功能,提高生存率,且安全性及耐受性较好,于2016年12月首次在国外获批使用,现已获得中国国家药品监督管理局正式批准,可用于治疗全年龄段的SMA患者。自2019年10月在国内应用以来,诺西那生已在全国25家医院用于61例患儿,初步疗效及安全性尚待评估。但该药需要鞘内注射给药,每4个月给药1次,终身治疗,费用非常昂贵。此外,首个治疗SMA的基因替代药物onasemnogene abeparvovec-xioi于2019年5月获得美国食品药品监督管理局批准,用于治疗2岁以下的SMA患儿,但尚未在我国获批上市,且其有效性及安全性仍需进一步研究。
除新型靶向治疗外,对SMA强调多学科综合管理,需要动态评估患儿的运动功能、呼吸功能、营养状况、脊柱侧弯和髋关节脱位等,并定期进行物理治疗、正确使用支具或矫形器、规律运动、功能训练、吞咽功能训练及呼吸功能训练等,积极的康复治疗仍是目前干预、延缓疾病进展的主要手段,应贯穿治疗全过程。对于晚期患儿应加强护理,预防肺部感染及压疮,如伴有严重呼吸功能不全,必要时需进行有创通气治疗,保证气道通畅,改善呼吸功能。
SMA以常染色体隐性方式遗传,在已明确诊断的SMA家庭中,每次怀孕都有大约25%的再发风险。由于SMA属于携带率高、发病率高的致残致死性疾病,产前诊断适用于所有存在生育SMA患儿风险的家庭,一般选择10~12孕周采集绒毛,18~22+6孕周采集羊水,以明确胎儿是否为患者、携带者或健康人。美国医学遗传学与基因组学学会建议对所有育龄人群实施SMN1携带者基因筛查,对高风险胎儿实施产前诊断或植入前诊断,从而减少SMA患儿的出生。
患儿父母多为SMN1单拷贝携带者,但需注意的是,约有4%的SMA携带者为“2+0”型(携带2个SMN1基因拷贝,但2个SMN1基因位于同一条染色体),对于这类携带者家庭的再发风险和产前诊断策略与SMN1单拷贝携带者相同。此外,约有2%的先证者可能在其中一个等位基因为新发SMN1突变,这种情况下仅有一个亲本为SMN1单拷贝携带者,而这几乎不会增加SMA患儿的再发风险。









