


 收藏
收藏 已收藏
已收藏英文名称 :hereditary spastic paraplegia
中文别名 :Strümpell-Lorrain病
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)又称为Strümpell-Lorrain病,是一组具有高度临床和遗传异质性的罕见的神经系统变性疾病,主要表现为缓慢进展的双下肢无力和痉挛性截瘫。HSP总体发病率为1~10/100 000,发病年龄差异较大,从新生儿到老年人都有。HSP的遗传方式包括常染色体显性遗传、常染色体隐性遗传、X连锁隐性遗传和线粒体遗传,其中常染色体隐性遗传最常见。
尽管HSP的遗传学表现多样,但共同的病理特征是皮质脊髓束和薄束后索轴突的长度依赖性变性,皮质脊髓束以胸段脊髓受累最重,薄束以颈段脊髓受累最重。双侧脊髓小脑束也有不同程度的病变,脊髓前角细胞、巨锥体细胞、基底节、胼胝体、小脑、脑干、大脑皮质和视神经也可累及。HSP涉及多种分子学病因,包括细胞内运输障碍、核苷酸代谢障碍、突触形成和轴突发育障碍、轴突运输障碍、线粒体障碍、髓鞘维护和组装障碍。
临床分型:HSP根据临床特征的不同分为单纯型和复杂型,单纯型表现为逐渐进展的双下肢痉挛、步态不稳、腱反射亢进,可以合并膀胱括约肌功能障碍,上肢也可能出现反射增强,但脑神经很少受损;复杂型除上述临床表现外还可伴有共济失调、严重的肌萎缩、视神经萎缩、视网膜色素变性、精神发育迟滞、锥体外系症状、智力障碍、耳聋、鱼鳞病、周围神经病和癫痫等;国外最常见的合并症状是肌萎缩,国内是智力障碍。
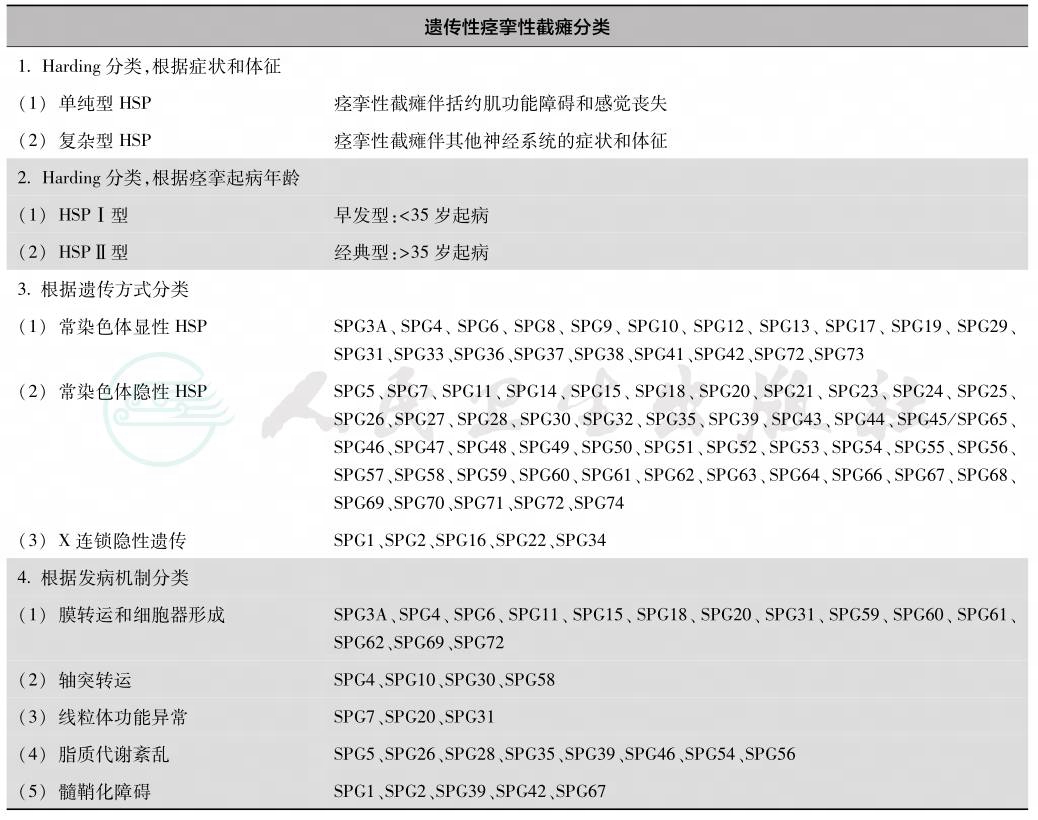
HSP基因型与临床表型:HSP的遗传分类基于其遗传方式、染色体位点和致病突变,其基因位点被命名为SPG(spastic paraplegia gene),并按顺序编号为 SPG1、SPG2、SPG3等,SPG编号是基因位点的发现顺序,而不是遗传传递机制,目前发现的基因位点数超过55个,并还在持续增加。HSP临床分类(单纯型或复杂型)与遗传分类(SPG型)之间的关联不完全,一些遗传类型与单纯型和复杂型均相关。随着分子遗传学诊断技术的提高,越来越多的HPS致病基因被发现,据EFNS指南报道的基因型与临床表型见表1。
表1 遗传性痉挛性截瘫分类及主要的亚型
本病发病机制不明。研究表明HSP的病理改变以轴索变性为主,可伴有脱髓鞘和神经元脱失等改变。退行性病变发生于脊髓的侧索及背索,尤其是这些纤维束的远端。受累最严重的脊髓胸段的皮质脊髓束和薄束纤维,而脊髓前后角细胞以及周围神经少有受累。HSP复杂型病变可累及其他神经结构、小脑、基底节、大脑皮层、白质以及胼胝体。
目前,HSP尚无有效治疗方法,以缓解临床症状,提高生活质量为主。传统的药物治疗有巴氯芬、替扎尼定、加巴喷丁、普瑞巴林等口服药物缓解肌张力,肌内注射肉毒杆菌毒素等防止挛缩和畸形。有报道鞘内注射巴氯芬和机器人步态训练能有效地改善单纯型HSP患者的平衡和行走能力。此外,还可考虑选择性外周神经切断术或其他外科手术,如软组织局部外科干预等治疗。









