


 收藏
收藏 已收藏
已收藏英文名称 :thalassemia
中文别名 :海洋性贫血
地中海贫血(thalassic anemia,缩写为thalassemia)因最初发现的病例都来自地中海地区而得名。也称海洋性贫血综合征(thalassemia syndrome)和珠蛋白生成障碍性贫血(anemia of imbalanced globin chain synthesis)。地中海贫血是一组遗传性异质性疾病,其中的每一种都是由一种遗传性珠蛋白合成异常所导致的,构成了被笼统称为血红蛋白病(hemoglobinopathies)疾病谱或血红蛋白遗传性异常疾病谱的一部分。血红蛋白病是世界上最常见和危害最严重的单基因遗传性疾病之一,大体上分为两类:第一类为珠蛋白链的遗传性结构改变导致的异常,如镰状细胞贫血、血红蛋白E病,这类血红蛋白病在我国少见;第二类为珠蛋白肽链一条或多条肽链合成缺陷所致的遗传性疾病,即地中海贫血,在我国多见。
地中海贫血主要有α、β地中海贫血两类,分别累及珠蛋白α、β链基因,少见类型是由其他珠蛋白基因异常所致。它们都有成人血红蛋白珠蛋白链的生成不平衡,即α地中海贫血因α链合成缺失或减少而β链相对过量;β地中海贫血因β链合成缺失或减少而α链相对过量。从分子水平看,是数百种α、β珠蛋白位点的突变而导致α、β链合成的缺失或减少。
地中海贫血是珠蛋白链基因中一个或一个以上缺失或突变,珠蛋白四条肽链中的某一条或一条以上的合成缺失或减少,导致血红蛋白的产生不充分,使红细胞呈现偏薄的低色素性小细胞和靶形(贫血与红细胞计数减少不相平行)特征,同时因不能匹配的珠蛋白链过剩(珠蛋白链之间合成比例上的不平衡)而沉积红细胞内,并引起红细胞氧溶解度下降或携氧能力下降,红细胞及其前期细胞损伤以及骨髓无效造血和溶血的遗传性异质性疾病。
(一)β重型地中海贫血
β重型地中海贫血(thalassemiamajor,TM),又称库利贫血,为β珠蛋白链合成受抑,其结果是不能形成成熟血红蛋白A,在1岁时即可发展到血红蛋白(2条α链和2条γ链)消失的程度,可以致命。多见于希腊和意大利小儿。常见临床症状有黄疸、肝脾进行性肿大、感染等。致死性心脏血红蛋白沉着病需要多种输血治疗来缓解贫血症状。室上性心律失常和慢性心衰也很常见。这些患者对洋地黄的作用是高度敏感的。由于骨髓长期显著增生,使骨髓腔变宽,骨皮质变薄。大量髓外造血和椎体破坏会造成血胸和脊索受压。上颌骨过度生长可能导致气管插管时使用直接喉镜难以暴露声门。
羟基脲对于一些镰状细胞β型地中海贫血有一定治疗作用(参见镰状细胞疾病)。对于这些患者应用骨髓移植有良好疗效。对有巨脾压迫腹腔脏器,行动不便者,有脾功能亢进,白细胞、血小板减少者,可行脾切除术。
(二)β轻型地中海贫血
β轻型地中海贫血(thalassemiaminor)染色体为杂合子状态,只导致轻度贫血。红细胞计数相对正常是β轻型地中海贫血与缺铁性贫血重要的鉴别点之一。这种类型的贫血比原来认识的更为常见。
(三)α地中海贫血
α珠蛋白链合成受抑者称为α地中海贫血。纯合子型α地中海贫血为重度致死性疾病,常胎死宫内或出生后不久即死亡。杂合子型α地中海贫血患者症状较轻,常为轻度的低血红蛋白或小细胞贫血。在一些情况下,可输血治疗或切脾以控制溶血。
几乎所有的地中海贫血的病理生理特点都与珠蛋白链的合成失去平衡有关。这一现象使得地中海贫血从根本上区别于其他遗传性和获得性血红蛋白合成疾病,而且可以在很大程度上解释纯合子和杂合子状态的严重性(图1)。
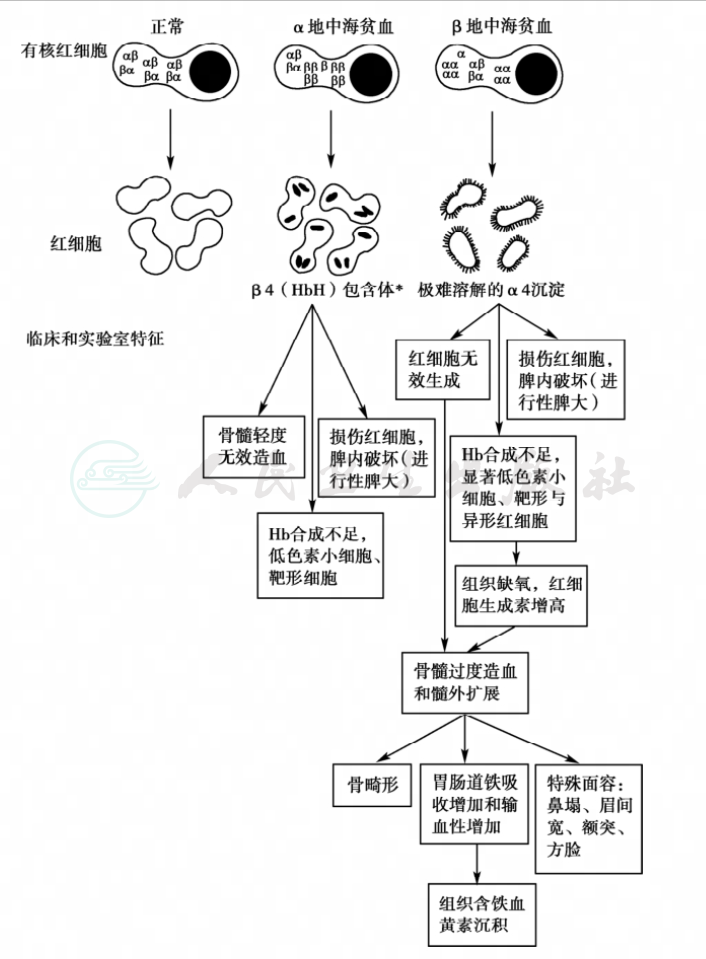
图1 地中海贫血的病理生理
*胎儿为γ4四聚体,产生Hb Bart









