


 收藏
收藏 已收藏
已收藏儿童HUS主要与感染相关,90%的HUS是由分泌志贺毒素的细菌感染所致,最主要的致病菌为大肠埃希菌(Escherichia coli,E)O157,常见于5岁以下儿童,但也可发生在<6个月的小婴儿。美国及西欧报道总体年发病率为0.2~2.1/10万,而小于5岁以下儿童年发病率为6/10万,畜牧业发展的国家可能更高,如阿根廷5岁以下儿童年发病率为10~17/10万[2-4]。本病常见于温暖季节,夏季常有小流行。感染常与食物污染及加工不当有关,特别是肉类、奶制品、家禽类食品,人和人之间也有传播可能。其他病原感染相对少见,如与肺炎链球菌感染相关者,0~17岁儿童的年发病率为0.015/10万[5]。在非感染病因所致HUS中,与补体旁路激活异常相关者最多见,占50%~60%,据报道20岁以下年龄的年发病率为0.26~0.75/百万,患病率为2.2~9.4/百万,0~4岁为最高峰,为 3/百万[1,6]。
中心环节是各种病因导致的微血管内皮细胞损伤。
1.STX-HUS [2,4,8]
一般感染了产 STX 的细菌后,引起腹泻,其中5%~15%发展为HUS。
(1)细菌毒素对细胞的毒性作用
STX由两个亚单位组成,一个32kd的A亚单位和一个五聚体B亚单位(每个7.7kd)。有两种主要的亚型:STX1和 STX2,两种STX的A和B亚单位分别有57%和60%的核苷酸序列同源,56%的氨基酸相同。因此,两种STX的生物学活性、受体结合力有显著差异,其中STX2与人类疾病更相关。
产STX的大肠埃希菌感染大肠后,引起局部致病过程,同时释放的STX穿过胃肠道上皮进入血液循环。STX通过B亚单位与血细胞中的红细胞、血小板、单核细胞膜上球丙糖基神经酰胺(globotriosylceramide,Gb3)受体结合,也可与中性粒细胞膜上的Toll样受体4(toll like receptor,TLR4)结合,转运至靶器官。与血细胞结合的STX也可内化,形成微泡并从血细胞中释放,到达靶器官。
到达器官的STX与微血管内皮细胞膜上的Gb3结合并内吞,或微泡直接被内皮细胞摄取,通过早期内涵体、高尔基复合体转运到内质网,在此处A亚单位与B亚单位分离,进一步 A亚单位被剪切为 A1(27kd)和A2,进入胞质。A1具有酶切作用,能使一种60S核糖体亚单位的28S核糖体核糖核酸(ribonucleic acid,RNA)在高度保守区的一个腺苷脱嘌呤,抑制核糖体功能,阻止蛋白质的合成,同时激活了核糖毒性应激反应,内质网内未折叠蛋白A1激活了未折叠蛋白应激反应,STX的B亚单位与靶细胞结合本身触发胞质转导级联反应。最终,触发了细胞一连串的促炎症、促血栓、促凋亡通路,导致内皮细胞抗黏附、抗炎、抗血栓特性的丢失,甚至凋亡。①激活重要的调节细胞因子/趋化因子基因表达的转录因子NF-κB和激活子蛋白-1,上调黏附分子如细胞间黏附分子-1(intercellular cell adhesion molecule-1,ICAM-1)、血管细胞黏附分子-1(vascular cell adhesion molecule-1,VCAM-1)及P选择素等和细胞因子/趋化因子如单核细胞趋化因子-1(monocyte chemoattractant protein-1,MCP-1)、白介素-1 和 8(interleukin-1,IL-1;interleukin-8,IL-8)及肿瘤坏死因子-α(tumor necrosis factorα,TNF-α)等,增加白细胞的募集、与内皮细胞的黏附及激活,同时,又反过来上调内皮细胞的Gb3受体表达,使内皮细胞更易与STX结合,增强STX对细胞的毒性作用。②表达血管性血友病因子(von Willebrand Factor,vWF)增多,促进血小板聚集,同时,STX还可直接减弱vWF裂解蛋白酶(a disintegrin-like and metalloproteinase with thrombospondin type 1 motif,ADAMTS13) 裂解vWF的能力,直接与血小板上Gb3受体结合激活血小板。③表达组织因子增多,活化凝血因子Ⅶ与内皮细胞结合,促进凝血酶的生成和纤维蛋白多聚体的合成。④使THBD从细胞表面脱落、丢失(THBD是使凝血酶由促凝转向抗凝的重要的血管内凝血抑制因子)。⑤大量STX时使内皮细胞凋亡和细胞脱离,暴露内皮下促血栓型组织因子和胶原。上述种种引发微血栓形成。
(2)细菌毒素引起补体旁路系统的异常活化
补体系统是一个由30多种蛋白组成的先天免疫系统,提供体液免疫和炎症中的许多效应功能,可介导细胞溶解、促进吞噬细胞对微生物的吞噬以及刺激白细胞释放作用于血管的炎性介质。补体激活途径有三种:经典途径、凝集素途径和旁路途径。经典途径通过抗原抗体复合物激活,凝集素途径由细菌表面的分子激活,而旁路途径则是C3自身活化,裂解为C3a(过敏毒素,促炎症因子)和C3b,C3b可沉积到任何与血浆接触的细胞表面,并与B因子相互作用产生Bb,C3b与Bb结合形成C3转换酶,持续激活补体C3,同时再与一个C3b结合形成C5转换酶,其裂解C5,促成攻膜复合物(MAC)的形成。MAC像一个细孔样结构插入细胞膜,引起细胞激活或溶解。正常情况下,为避免旁路途径过度活化攻击自身细胞,体内存在众多补体调节蛋白对其活化进行精细调节,包括血浆中和细胞表面的一些能促进C3b裂解为无活性iC3b,分解C3/C5转换酶,阻止C9组装C5b-9形成的因子,如血浆中的补体H因子、I因子,膜结合调节蛋白CD46又称膜辅因子蛋白(membrane cofactor protein,MCP)、CD55又称衰变激活因子(decay activating factor,DAF),CD59又称膜攻击复合物抑制因子(membrane attack complex inhibitory factor,MACIF) 等(图1)。
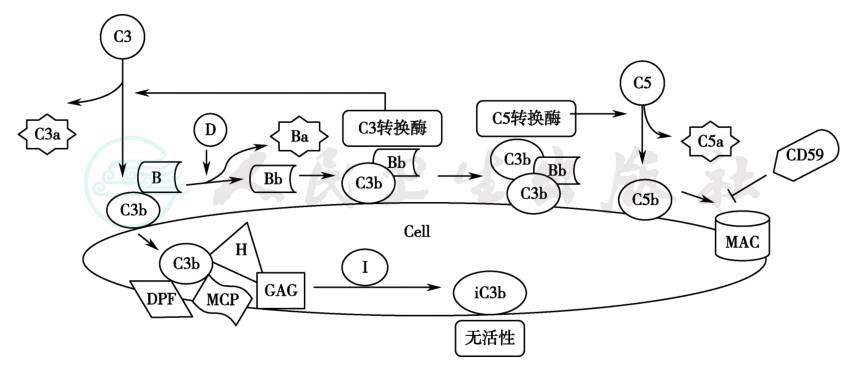
图1 补体旁路途径激活及调节
在STX-HUS中,补体旁路途径激活参与致病的临床证据:该类患儿可出现血补体C3降低,C3分解产物(C3b、C3a、C3d)、血浆 Bb 及可溶性 C5b-9水平升高,肾脏C3和C5b-9沉积,含STX的血细胞微泡被覆补体成分C3和/或C9,一起被转运到靶细胞,28%的STX-HUS儿童有血清学或基因学上的补体异常。此外,抗C5单克隆抗体eculizumab成功治疗STX-HUS的患儿,也从临床实践的角度进一步证实了补体活化在STX-HUS发病中的地位。
研究表明:①STX能直接激活补体:STX2直接结合H因子和具有类似功能的H因子家族中的FHR-1、FHL-1,使其不能在细胞表面起到辅助因子的作用,导致补体激活增加,C3b堆积;STX还能减少CD59表达。②STX引起内皮细胞表达增多的P选择素可与C3b结合,激活补体旁路途径。③STX导致THBD从细胞表面丢失,同样使补体旁路激活(正常情况下THBD灭活C3a、C5a,加强I因子介导的C3b灭活)。④STX可激活补体凝集素途径,通过C4/C2旁路机制放大补体旁路的激活。
补体旁路激活在STX-HUS的微血栓形成中起关键调节作用:①补体激活成分C3a、C5a与内皮细胞膜上相应受体C3aR、C5aR结合以及C5b-9的形成,均具有促炎症活性,诱导黏附分子上调和细胞因子分泌,加之C3a、C5a本身趋化和激活的特性,加重白细胞聚集、黏附、激活和内皮损伤。②C3a与其内皮细胞C3aR结合,增强P选择素表达及THBD的脱落丢失。③B因子及补体旁路激活促进血细胞的微泡释放。
(3)肾脏是HUS主要受累器官的机制
目前尚不十分清楚。首先肾脏血流丰富,增加了毒素暴露的机会。其次Gb3受体在体内的分布范围,决定了微血管病变的部位,受体密度决定了该细胞对STX的易感性,而肾小球毛细血管内皮细胞、系膜细胞和肾小管上皮细胞的细胞膜上Gb3受体数目均较多,特别是儿童肾脏Gb3表达比成人还多,可能导致儿童肾脏最易受STX的侵袭。此外,肾小球毛细血管结构特殊,使得表达Gb3的足细胞也是STX的靶细胞,在STX-HUS患儿尿中发现足细胞特征性标志mRNA,提示足细胞确实被STX损害。STX可激活足细胞表达IL-1和TNF-α,上调Gb3,增加其对STX的敏感性;增加足细胞血管活性肽内皮素-1产生,引起足细胞骨架蛋白重排,改变肾小球血流动力学;减少VEGF表达,使内皮细胞功能异常,减少局部H因子的活性。循环和局部产生的C3a与足细胞相应受体结合,使骨架蛋白α-actinin-4表达减少,nephrin下调,导致足细胞功能紊乱、脱落。
2.肺炎链球菌相关HUS(Streptococcus pneumoniae related HUS,Sp-HUS) [3,5]
是侵袭性肺炎球菌感染罕见但严重的并发症,首次由Fischer于1971年描述。Sp-HUS机制尚不明确,Thomsen-Friedenreich(TF)抗原可能起关键作用。链球菌产生一种链球菌毒素N-乙酰神经氨酸酶(N-acetylneuraminidase)又叫唾液酸酶(sialidase),裂解红细胞、血小板及肾小球内皮细胞膜上的各种糖蛋白、糖脂的N-乙酰神经氨酸,使细胞膜表面暴露TF隐蔽抗原,该抗原特异性地被花生植物血凝素识别,引起RBC凝集,导致溶血;并与其相应的IgM抗体发生反应,导致内皮细胞损伤继而出现血管内凝血。此外,由于细胞表面唾液酸的裂解,使补体H因子不能发挥调解补体旁路的作用,使补体激活,最终HUS发生。
3.补体介导aHUS [3,8,10]
自 1998 年 Warwicker报道H因子基因突变导致aHUS,现已发现120种以上的补体旁路调节基因突变与aHUS相关。
(1)携带与补体旁路途径相关蛋白基因突变
①H因子基因突变:最常见,检出率为20%~30%。H因子是旁路途径的主要调控因子,是I因子的辅助因子,也是与B因子竞争性结合C3b的加速衰变因子。目前在散发和家族性aHUS中已发现100种以上不同的突变,主要是杂合突变,多集中在C端的 C3b结合位点SCR19-20,突变虽然不影响血H因子水平,但与配体(C3b、氨基多糖、肝素)结合能力下降。此外,H因子相关蛋白1~5(complement factor H-related protein,CFHR 1~5)的基因紧邻H因子基因,CFHR 3~5具有辅助因子活性,CFHR 1~2可抑制C5转换酶、C3转换酶形成。临床上常见的基因异常为CFHR与H因子基因发生非等位基因同源重组的融合基因出现,产生的杂交H因子功能丢失。目前已报道与aHUS相关的6种杂交类型。②CD46(MCP)基因突变:见于8%~10%的 aHUS。MCP是I因子介导裂解C3b的辅助因子,现已发现40种突变,多数为杂合突变,集中位于MCP细胞外C3b结合位点,导致MCP表达下降,与C3b结合减弱和辅助因子活性减低。③I因子基因突变:见于4%~10%的aHUS。I因子是重要的补体旁路调节因子,在特定的辅助因子H和MCP存在下裂解C3b。已发现的40种突变均为杂合突变,主要位于编码丝氨酸蛋白酶的外显子,导致I因子水平减低或蛋白水解活性异常。④C3基因突变:见于4%~8%的 aHUS,但日本报道高达31%。C3是肝脏合成的关键补体蛋白,多为杂合突变,主要位于H因子结合位点,使C3与H因子、MCP的结合减低,导致I因子介导的C3b灭活受损,属于功能获得性突变。少见的C3p.R139W突变增加C3与B因子的结合,形成高活性的C3转换酶。⑤B因子基因突变:见于<1%~4%的aHUS。目前已发现4种杂合突变,也属于功能获得性突变,使B因子过度与C3b结合,增强C3转换酶的稳定性和活性,血C3非常低。
研究发现在相关补体突变基因的携带者中,aHUS的外显率约是50%~60%,即家族中携带补体基因突变者仅部分人发生aHUS,提示aHUS的发生和进展必须存在其他的基因修饰(常见H和CD46基因多态性修饰其风险)和环境因素的调控,即一种以上补体因子的基因突变或与风险增加的多态性突变同时存在,在aHUS发展中起重要因素(多重打击学说)。
(2)获得性旁路途径补体调控缺陷
获得性H因子自身抗体相关aHUS,占aHUS的5%~20%,印度报道更高,占56%。多见于5~10岁儿童。抗H因子抗体IgG与H因子SCR19和20位点结合,抑制了H因子与C3在细胞表面的结合,从而抑制了H因子对补体旁路途径的调控作用。该抗体本身还可导致绵羊红细胞溶解,溶解程度与抗体滴度相关,可受H因子抑制。多数抗H因子阳性患儿存在CFHR1基因纯合缺失,有时也可见CFHR3或CFHR4纯合缺失,其导致自身抗体出现的机制不清,推测:H因子存在自身抗原表位,当微生物分子靠近该区域结合时,其位点被表达,CFHR1蛋白也存在一个结构类似的自身抗原性的表位,起到免疫耐受作用。当缺乏CFHR1而免疫耐受丢失时,导致自身免疫性aHUS发生。
4.非补体介导aHUS [3,10]
一些非补体系统的蛋白基因突变,同样也有aHUS的表现。①THBD基因突变:2009年首先被报道,见于3%~5%的aHUS。THBD为抗凝血糖蛋白,降低凝血酶的凝血活性、增加其激活蛋白C的活性,同时可灭活C3a和C5a、促进I因子介导C3b灭活。常见为杂合突变。②DGKE基因突变:2013年首先被报道,见于8%的患者,有纯合突变及复合杂合突变。DGKE是脂质激酶家族蛋白,使花生四烯酸甘油二酯磷酸化为磷脂酸,终止其介导的蛋白激酶C信号通路激活(该信号通路激活可促进凝血酶诱导的血小板激活及促凝因子的释放),具有抗凝血作用。③PLG基因突变:目前报道了4例患者。PLG是纤维蛋白溶酶的前体,降解纤维蛋白凝胶,阻止血小板聚集,并可激活C3、C5。④M MACHC基因突变:MMACHC参与维生素B12又称钴胺素(cobalamin,Cb1)在细胞内的代谢,其基因突变使Cb1不能转化为甲基钴胺素(蛋氨酸合成酶的辅酶,将同型半胱氨酸转为蛋氨酸)和腺苷钴胺素(甲基丙二酰辅酶A变位酶的辅酶,使甲基丙二酸单酰辅酶A转变为琥珀酰辅酶A,将甲基丙二酸转为琥珀酸,参与三羧酸循环),引起伴高胱氨酸尿症的甲基丙二酸尿症,内皮细胞损伤。25%表现有aHUS,或为首发症状。⑤其他:新近发现引起家族性常染色体显性遗传肾病综合征的反式甲酸2(inverted formin 2,INF2)基因突变,可诱发aHUS,阻断补体激活的药物依库利珠单抗(eculizumab)治疗无效,其机制不清。
目前尚未统一。随着对HUS认识的深入,按病因学分型如下[3,7-9]:
1.感染相关性HUS(infection-associated HUS)与产志贺毒素的细菌感染相关,如大肠埃希菌、痢疾杆菌或空肠弯曲杆菌,称之为志贺毒素型HUS(Shiga toxin HUS,STX-HUS),也是以往所说的典型 HUS(typical HUS)或腹泻相关型 HUS(diarrhea related HUS,D+HUS)。其中大肠埃希菌EO157:H7最多见,产STX毒力强,其他产 STX 的菌型还有 O26、O55、O80、O91、O103、O111及 O104:H4等,均可引起流行,近年来非O157型感染有增多趋势。此外,尚有肺炎链球菌、肺炎支原体、流感病毒、诺如病毒、HIV等感染所致HUS。
2.不典型HUS(atypical HUS,aHUS)过去特指与补体系统相关蛋白基因突变或获得性自身抗体产生导致补体旁路异常活化所致的HUS。随着基因学检测的快速发展,发现表现为aHUS的患者中还有其他非补体系统相关蛋白基因突变所致者,故又将aHUS分为补体介导 aHUS(complement-mediated aHUS)和非补体介导aHUS(non-complement mediated aHUS)。 后者如目前已报道的甘油二酯激酶(diacylglycerol kinase ε,DGKE)、血栓调节蛋白(thrombomodulin,THBD)、血纤维蛋白溶酶原(plasminogen,PLG)及甲基丙二酸尿和同型胱氨酸尿C蛋白(methylmalonic aciduria and homocystinuria type C protein,MMACHC)基因突变所致的aHUS。
3.继发性HUS(secondary HUS)见于:①自身免疫性疾病:如系统性红斑狼疮、抗中性粒细胞胞质抗体相关性血管炎、抗磷脂综合征、硬皮病、IgA肾病、C3肾病等。②药物治疗:化疗药如奎宁、丝霉素、顺铂、吉西他滨;免疫抑制药如环孢素、他克莫司、西罗莫司;抗血小板药如氯吡格雷、噻氯匹定;抗生素如青霉素、环丙沙星、磺胺异𫫇唑;血管内皮生长因子(vascular endothelial growth factor,VEGF)和酪氨酸激酶抑制剂如贝伐单抗、阿柏西普、舒尼替尼、索拉菲尼、西地尼布;其他如干扰素、伐昔洛韦、电离辐射、口服避孕药等。③肿瘤,如胃癌、乳腺癌、肠道肿瘤及血液恶性疾病等。④恶性高血压。⑤妊娠。⑥实体器官和骨髓移植。
4.不明原因的HUS。
以多脏器微血管病变、微血栓形成为其特点,肾脏受累最重,其次为肠、脑、胰、心、脾、肾上腺等器官。急性期可见不同程度的肾小球及小动脉病变,包括内皮细胞肿胀,内皮细胞与基底膜分离,致毛细血管外周袢呈“双轨样”改变或明显分层;可伴节段袢坏死,毛细血管腔内可见微血栓及血小板,导致袢腔狭窄或完全阻塞。肾小管上皮细胞变性、坏死,肾间质水肿,可见单核细胞浸润。叶间动脉及入球小动脉往往受累,小动脉血栓形成,内皮细胞增殖呈“洋葱皮样”向心性增厚,当管腔狭窄或完全闭锁时,肾小球毛细血管袢塌陷、基底膜增厚皱缩,呈不同程度的缺血样改变。严重者可导致肾皮质坏死。病变晚期可见肾小球硬化、玻璃样变、肾小球荒废、肾小管萎缩及间质纤维化。免疫荧光:急性期肾小球毛细血管袢常见纤维素/纤维蛋白相关抗原沉积,有的病例可见IgM、C3分布于外周袢,IgG少见,罕见IgA沉积。间质动脉和小动脉壁和/或内皮下亦可见纤维素/纤维蛋白相关抗原沉积。此外,尚可见IgG、C3、C1q等阳性。肾小球毛细血管袢内及动脉和小动脉内的血栓亦可见纤维素/纤维蛋白相关抗原阳性。
1.血常规 由于急性溶血使血红蛋白迅速下降至70~90g/L,严重者达30~50g/L;90%病例血小板减少,可低至10×109/L,通常在1~2周内恢复正常,部分患儿血小板无减低,被称为不完全型HUS;白细胞升高可达20×109/L以上,以中性粒细胞为主,和预后有一定关系。
2.血管内溶血相关指标 末梢血片中可见形态异常的破碎红细胞,呈三角形、盔甲形、芒刺状等,网织红细胞增高,结合珠蛋白阴性,血乳酸脱氢酶、磷酸激酶、转氨酶及胆红素升高。
3.凝血功能 凝血酶原时间、部分凝血活酶时间正常,纤维蛋白降解产物升高,纤维蛋白原正常范围。
4.尿常规及肾功能 患儿几乎都有血尿及轻重不等的蛋白尿、氮质血症、高钾血症、低钙血症及代谢性酸中毒等,且随少尿而加重。
5.Coombs试验 阴性,但Sp-HUS绝大多数直接Coombs阳性。
6.血补体C3 部分STX-HUS患儿C3减低。补体介导aHUS约30%~80%减低,特别是H因子纯合突变、补体C3及B因子基因突变者血C3水平非常低,而MCP基因突变绝大多数C3正常。
7.补体旁路及其他相关基因学检测、抗H因子抗体检测。
8.病原学检测 感染相关的HUS根据不同病因进行便、血等细菌培养,粪便STX检测,血相关抗体检测等,明确感染源。
9.肾活检 必要时。
1.支持疗法
(1)维持机体水电解质平衡及营养支持
研究表明患儿腹泻的4天内静脉补充足够的液体,补充累积损失及继续损失,可减轻AKI,降低透析的风险。不能进食或腹泻严重者应给予胃肠道外营养支持,以免加重氮质血症或出现严重低蛋白血症。另外,如果患儿有明显水肿、少尿及高血压,应限制水、钠入量,可酌情用利尿剂呋塞米,每次2mg/kg,血钾偏高应限制钾入量,一旦血钾>6mmol/L应紧急处理。
(2)控制高血压
一般用硝基苯吡啶口服,每次0.25~0.5mg/kg(每次<10mg),必要时硝普钠每分钟0.5~8ug/kg静脉输注,急性期后的高血压可用ACEI类降压药。
(3)控制惊厥发作
可用地西泮每次0.2~0.3mg/kg,缓慢静脉注射。
(4)纠正严重贫血
当血细胞比容下降到15%或Hb<70g/L时,可输注新鲜红细胞悬液5~10ml/kg。一般避免输血小板,因它可能加重微血栓,但如有严重活动性出血及外科或插管操作时需要输注。Sp-HUS如病情需要应选择洗涤的红细胞或血小板。
(5)透析疗法
约50%的HUS患儿需要透析治疗,Sp-HUS约75%~100%需要透析治疗,是降低急性期病死率的关键方法之一。凡无尿大于24小时,氮质血症,尿毒症,对利尿剂无反应的严重液体负荷(心衰、肺水肿及顽固高血压),对药物治疗无效的高血钾(血钾>6.5mmol/L)和酸中毒(pH值<7.1)等都应尽早开始透析治疗。
对婴幼儿一般采用腹膜透析,对有严重结肠炎或腹膜炎者需采用血液透析或超滤。透析不能改变疾病的病程,但能纠正液体、电解质紊乱及营养支持,等待疾病的缓解。
2.血浆置换
是aHUS的一线治疗[1]。新鲜冰冻血浆(fresh frozen plasma,FFP)不但能补充正常的 H、I、B、C3等成分,经过置换还能去除突变的补体成分、抗H因子抗体及可能存在的参与内皮损伤、促进血小板凝聚的炎症和血栓形成因子。一经诊断aHUS,尽快开始,FFP 1.5倍血浆量或60~75ml/kg,每天一次(至少 5天)至血小板正常、溶血停止(血乳酸脱氢酶和血红蛋白正常)及肾功能持续改善几天后,逐渐减少置换次数,根据病情几周或几个月后停用。常见血浆置换的并发症有管路阻塞、低血压及过敏。
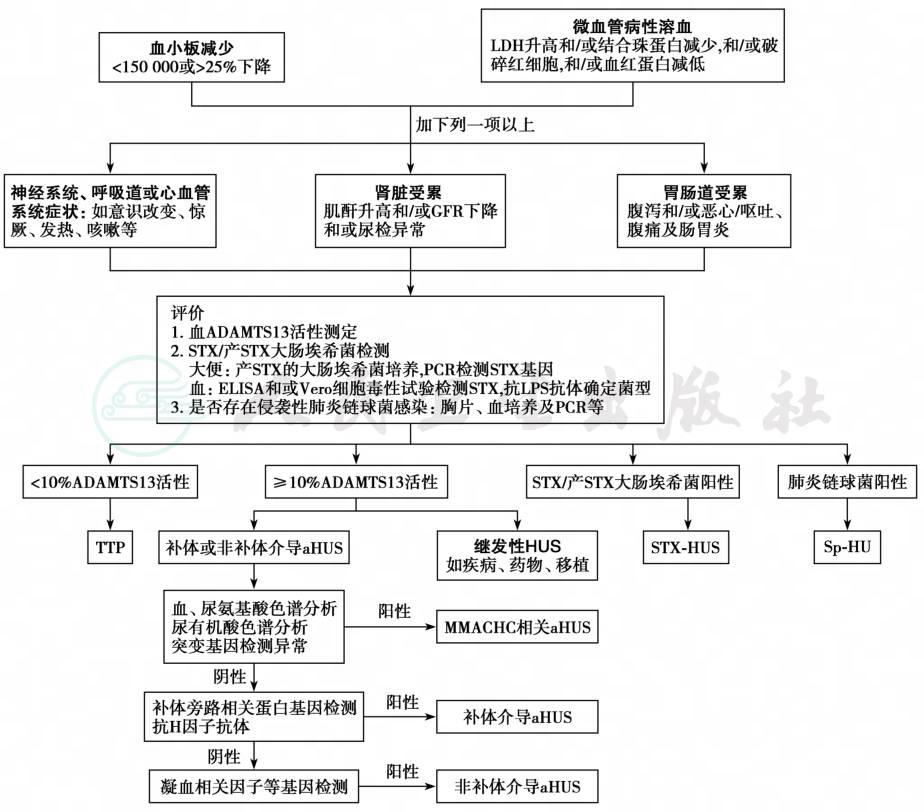
图2 HUS的临床诊断流程
如即刻没有条件进行血浆置换,可给予FFP输注10~20ml/kg,但应注意过敏、容量负荷过高、高血压、心衰及高蛋白血症等并发症。
因MCP是膜蛋白,不是循环中的血浆因子,故MCP基因突变患儿血浆置换无明显效果。对STX-HUS的益处证据也不足,但有些中心仍报道对严重危及生命的患儿有效[2,11],推测其可补充前列腺素 I2(prostaglandin I2,PGI2)生成刺激因子、补充PGI2及补充其他抑制血小板聚集的因子,去除某些促炎症及血栓因子或毒素结合的抗体。对Sp-HUS是否给予血浆置换,也存在争议,因为成人血浆中含有针对TF抗原的IgM抗体及神经氨酸酶,故多数学者推荐避免应用,但临床中罕见给血浆或未洗涤RBC后病情加重的报道,而且血浆置换能够去除抗TF抗体和血浆神经氨酸酶活性,对重症Sp-HUS治疗可能有效[5]。对严重的新生儿或小婴儿Sp-HUS,尚可进行换血治疗,不但能够清除循环中的抗TF抗体和神经氨酸酶,还能清除TF暴露的RBC。
3.阻断补体激活
依库利珠单抗(eculizumab)是一种抗C5单克隆抗体,与C5特异性结合,阻断其分解为C5a和C5b,从而阻止攻膜复合物C5b-9的形成,目前已作为补体介导aHUS的最佳有效治疗方法,85%可达缓解,也被推荐为一线治疗方案[1],对有补体系统参与致病的STX-HUS及Sp-HUS等重症病人,也有有效的报道[2,3]。不同体重儿童 eculizumab的推荐方案见表1。但由于aHUS易复发,具体eculizumab需要维持用药多长时间目前尚无统一意见,该药主要的副作用为增加感染奈瑟菌脑膜炎球菌的机会,一方面建议用药前给予疫苗注射和/或青霉素、阿莫西林等抗生素预防,另一方面建议病情控制并完全恢复后逐渐停药,复发时再次给予治疗,从而减少持续用药感染的风险。
表1 不同体重儿童eculizumab的推荐方案

注:如果与血浆置换或血浆输注联合治疗时,则每次血浆置换后的60分钟内再给予300mg或600mg,或每次血浆输注前60分钟内再给予300mg/每单位血浆。
4.免疫抑制治疗
H因子抗体阳性患儿同时还应加用免疫抑制剂如糖皮质激素联合环磷酰胺或吗替麦考酚酯或抗CD20单抗等治疗,有助于改善预后。此外,基于STX能导致炎症性细胞因子增加,补体旁路活化,STX-HUS应用肾上腺皮质激素是有理由的,一项随机对照研究确实显示应用肾上腺皮质激素组血肌酐水平下降快于不用组,但目前尚未推荐应用。
5.其他
①产STX细菌感染时是否应用抗生素尚有争论,有报道显示抗生素不增加HUS的风险,也有许多报道显示风险增加,因其能刺激毒素的释放,增加及加重HUS的危险。研究显示应用抗生素的时间及种类与HUS有关:β内酰胺类和磺胺异𫫇唑-甲氧嘧啶增加HUS风险;而磷霉素最有可能减少HUS的发生,特别是在腹泻发生后3天内接受治疗;喹诺酮类也有降低HUS发生的风险,黏菌素、庆大霉素和利福平在体外实验中显示有效,但尚需临床进一步评估[13]。②Sp-HUS时感染也较重,可能危及生命,需要充分抗感染治疗,随着青霉素耐药的增多,在缺乏药敏试验的情况下经验性选择三代头孢菌素和万古霉素治疗。早期应用特异性神经氨酸酶中和抗体IVIG治疗可能会明显改善预后。胸腔积液和脓胸时,给予充分引流。③MMACHC相关的aHUS应尽早肠道外羟钴胺素治疗,并加口服肉碱、叶酸等药物。
6.肾移植
HUS导致ESRD可考虑肾移植。但aHUS 因复发率较高[1,3,11],导致移植物丧失的发生率也高,其中 H因子基因突变者移植后复发率最高,达68%~90%,而MCP基因突变者比较低,在10%~20%(移植后的肾脏可表达MCP)。供者可能没有被检测到补体突变,可能促进aHUS发生,故尽可能避免活体肾移植。随着基因检测技术的进步和eculizumab的应用(在移植前首先明确患儿的突变基因,并系统全面地进行供者的补体系统基因分析及预防性给予eculizumab),大大提高了活体肾移植的成功率。此外,因为H、B及I因子在肝脏中合成,故尚有肝肾联合移植的报道。
[1]LEE H,KANG E,GYUNG KANG H,et al.Consensus regarding diagnosis and management of atypical hemolytic uremic syndrome.Korean J Intern Med,2020,35(1):25-40.
[2]JOSEPH A,COINTE A,MARIANI KURKDJIAN P,et al.Shiga Toxin-Associated Hemolytic Uremic Syndrome:A Narrative Review.Toxins,2020,12(2):67-111.
[3] SHEERIN NS,GLOVER E.Haemolytic uremic syndrome:diagnosis and management.F1000Research,2019,8(F1000 Faculty Rev):1690-1700.
[4]BUELLI S,ZOJA C,REMUZZI G,et al.Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome.Microorganisms,2019,7(1):15-30.
[5]GUERRA OJL,RODRÍGUEZ RSG,CAMACHO WJM, et al.Hemolytic uremic syndrome associated with streptococcus pneumoniae in pediatrics:a case series.rev paul pediatr,2020,38:e2018065.
[6]YAN K,DESAI K,GULLAPALLI L,et al.Epidemiology of Atypical Hemolytic Uremic Syndrome:A Systematic Literature Review.Clinical Epidemiology,2020,12:295-305.
[7]AIGNER C,SCHMIDT A,GAGGL M,et al.An updated classification of thrombotic microangiopathies and treatment of complement gene variant-mediated thrombotic microangiopathy.Clin Kidney J,2019,12(3):333-337.
[8]BOWEN EE,COWARD RJ.Advances in our understanding of the pathogenesis of hemolytic uremic syndromes.Am J Physiol Renal Physiol,2018,314(3):F454-F461.
[9]BRUYAND M,MARIANI-KURKDJIAN P,LE HELLO S,et al.Paediatric Haemolytic Uraemic Syndrome Related to Shiga Toxin-Producing Escherichia coli,an Overview of 10 Years of Surveillance in France,2007 to 2016.Euro Surveill,2019,24(8):1800068-1800076.
[10]YOSHIDA Y,KATO H,IKEDA Y,et al.Pathogenesis of Atypical Hemolytic Uremic Syndrome.J Atheroscler Thromb,2019,26(2):99-110.
[11]CANPOLAT N.Haemolytic uremic syndrome.Turk Pediatri Ars,2015,50(2):73-82.
[12]ZINI G,DE CRISTOFARO R.Diagnostic Testing for Differential Diagnosis in Thrombotic Microangiopathies.Turk J Hematol,2019,36(4):222-229.
[13]KAKOULLIS L,PAPACHRISTODOULOU E,CHRA P,et al.Shiga toxin-induced haemolytic uraemic syndrome and the role of antibiotics:a global overview.J Infect,2019,79(2):75-94.









