


 收藏
收藏 已收藏
已收藏英文名称 :facioscapulohumeral muscular dystrophy
中文别名 :Landouzy‐Dejerine型肌营养不良症;相对轻度局限性肌营养不良
面肩肱型肌营养不良症(FSHD)(MIM:158900,158901)是仅次于假肥大型肌营养不良症、强直性肌营养不良症的第三位最常见的肌营养不良症,以不对称性面部肌肉、肩胛带肌和上臂肌群进行性肌萎缩、肌无力为典型临床特征;发病率为1/2万~1/1.5万,约95%患者呈常染色体显性遗传,通常具有遗传早现现象。FSHD从婴儿到老年均可发病,多数患者在10~30岁时发病,但约10%的患者可在5岁前发病;除上述肌肉受累外,病情进展还可累及躯干肌和下肢肌,还可出现高频听力下降、视网膜血管病变、呼吸受累、癫痫和智力发育迟滞等。
FSHD是遗传因素与表观遗传因素共同作用的疾病。FSHD 1型和FSHD 2型临床上无法区分,分子遗传变异机制不同,但目前倾向于认同两者都是由于D4Z4重复序列DNA甲基化水平降低,在表观遗传效应调控下染色质构象失去稳定性,引起转录抑制状态的DUX4(double homeobox4)基因在骨骼肌细胞中异常表达,产生的DUX4蛋白对肌细胞产生多种毒性作用,最终导致骨骼肌细胞的凋亡和萎缩、炎症反应和氧化应激等。如图1。
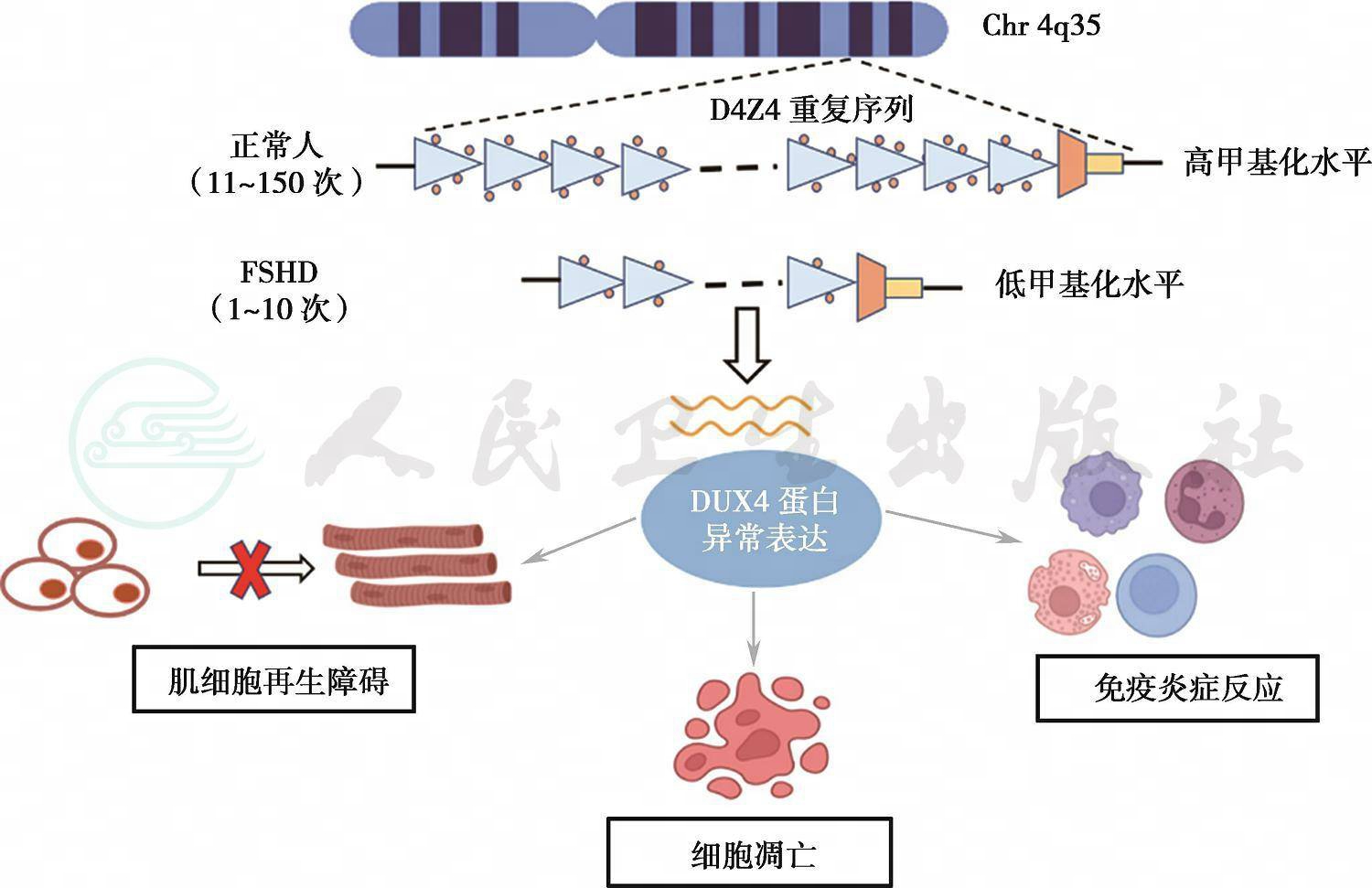
图1面肩肱型肌营养不良症(FSHD)发病机制模式图
引自:临床神经遗传病学.第1版.ISBN:978-7-117-33759-5.主编:
DUX4基因位于D4Z4重复序列中,包含2个同源序列和2个富含鸟嘌呤-胞嘧啶(GC)的重复序列,其末端连接稳定DUX4基因转录和翻译的多聚腺苷酸信号。DUX4基因编码2条全长(DUX4-f1)和截短(DUX4-s)的DUX4蛋白,在人类生殖细胞和早期胚胎干细胞中正常表达,DUX4基因在胚胎早期对诱导合子基因组激活起关键调节作用,此后则处于沉默状态,但在FSHD患者骨骼肌中呈异常表达。DUX4-f1蛋白是一个转录激活因子,可以引起下游多种改变,激活一系列去抑制级联反应,导致肌细胞病理生理学变化,但其具体生物学功能尚未完全阐明。目前较为公认的DUX4蛋白在FSHD中的病理生理学机制包括:①细胞凋亡学说,DUX4蛋白可以诱导p53抑癌基因表达,导致细胞凋亡,引起肌肉损害;②T淋巴细胞介导的细胞炎症反应学说,DUX4蛋白异常表达可以激活免疫反应,激活CD4+ T细胞和CD8+ T细胞,发生以T淋巴细胞介导为主的血管周围炎性细胞浸润,引起肌细胞肥大和细胞核聚集,导致肌肉损害;③DUX4蛋白异常表达可抑制肌细胞分化与再生。
D4Z4重复序列富含CpG岛,CpG岛主要位于转录调控区附近,是一种重要的表观遗传学修饰元件。正常人的D4Z4拷贝数大于10次,且呈高度甲基化,而FSHD 1型患者D4Z4拷贝数缺失,为1~10次。FSHD患者的D4Z4重复序列内存在3个DNA低甲基化区域,即DR1、DR2和DR3区域,其DNA甲基化总体水平显著低于正常人,尤其以DR1区域(位于D4Z4基因5’端)甲基化程度最低。DNA甲基化降低可引起局部DNA构象稳定性改变,从而导致毒性DUX4蛋白异常表达。
SMCHD1基因是FSHD 2型的致病基因,编码的蛋白通过CpG岛DNA甲基化的作用调控失活型X染色体和常染色体上的转座基因的染色质抑制。SMCHD1基因致病性突变与D4Z4重复序列低甲基化(DNA甲基化<25%,在正常人中约为50%)呈现共分离,SMCHD1蛋白可以直接结合到D4Z4重复序列上,通过甲基化修饰的作用抑制DUX4基因的表达。SMCHD1基因致病性突变可激活DUX4基因转录,从而增加DUX4蛋白的表达。SMCHD1基因致病性突变引起FSHD 2型的可能机制为单倍剂量不足,即SMCHD1蛋白水平减少使D4Z4重复序列的甲基化程度减低,从而导致DUX4基因的病理性表达。另外,SMCHD1基因致病性突变既可以引起FSHD 2型,也是FSHD 1型疾病严重程度的影响因素。研究发现DNMT3B、LRIF1基因也与FSHD 2型相关。
4q35和10q26区域均存在D4Z4重复序列,根据单体型的不同分为4qA、4qB、10qA、10qB四种类型,且四者序列高度同源,但只有4qA片段能够稳定表达DUX4基因,是FSHD的重要遗传学基础。
FSHD根据遗传方式可分为两型,即FSHD 1型和FSHD 2型,其中,FSHD l型占95%,呈常染色体显性遗传;FSHD 2型占5%,遗传方式多样,可呈常染色体显性或隐性遗传,临床上难以区分两型。
FSHD 1型:①位于4q35区域的D4Z4重复序列缩短,患者的拷贝数为1~10次(正常人的重复次数超过10次);②缩短的D4Z4重复序列位于4qA单体型上;③D4Z4重复序列甲基化水平显著降低。
FSHD 2型:D4Z4重复序列拷贝数正常,但甲基化水平显著降低,靶向测序可发现DNA甲基化调控基因SMCHD1基因、DNA甲基转移酶3B基因DNMT3B基因或配体依赖的核受体相互作用因子1基因(LRIF1)突变。
FSHD基因诊断方法包括:基于EcoR I+Bln I双酶切基因组DNA脉冲场凝胶电泳或琼脂糖凝胶电泳联合P13E-11探针的Southern印迹杂交技术,或基于单分子荧光原位杂交(FISH)技术;前者可通过完整分离4q和10q同源性EcoR I区域的全部片段,从而直观分析各种复杂易位带型并判断体细胞嵌合,但由于该技术方法操作复杂,限制了其临床应用;后者可使D4Z4重复序列可视化,通过不同荧光标记探针单次检测即可区分4q或10q来源,以及4qA或4qB单倍体型,并判断细胞嵌合。
D4Z4拷贝数与FSHD 1型的疾病严重程度呈负相关,D4Z4拷贝数越少,发病年龄越早,疾病进展越快,越可能失去行走能力及出现骨骼肌以外的症状;如存在1~3个D4Z4拷贝数的患者,发病早、进展更快、运动功能受累更严重,常伴有视网膜病变、感音性耳聋、癫痫等并发症;如存在7~10个D4Z4拷贝数的患者,临床表型轻微或为无症状携带者。除D4Z4拷贝数对临床表型的影响外,甲基化水平亦与患者的临床表型相关,甲基化水平越低,患者的临床表型越严重。
FSHD的病理改变多样,呈非特异性慢性肌病改变,无特异性病理特征;部分患者仅有轻度肌源性损害,甚至无明显肌肉异常。受累肌肉肌内膜、肌间质血管周围可见炎症细胞浸润(特别是单核细胞炎症反应),跨壁血管炎;肌纤维大小不一致,可见肌纤维变性坏死、纤维增生;晚期可出现脂肪组织和结缔组织明显增生。NADH-TR染色可观察到肌原纤维网格状结构排列紊乱。电镜主要表现为线粒体肿胀、坏死,肌细胞膜锯齿状改变。
1)实验室生化检查:
FSHD为肌纤维膜非破坏性肌肉病,血清肌酸激酶、乳酸脱氢酶水平正常或轻度升高,但极少超过正常值上限水平的5倍。
2)神经电生理检查:
肌电图呈肌源性损害,为运动单位动作电位时限缩短、波幅降低;部分患者短棘波多相电位增多,最大收缩呈病理干扰相或早募集。
3)神经影像学检查:
肌肉MRI检查可发现受累肌肉异常,或临床症状出现之前发现受累肌肉异常,可作为临床严重程度的预测指标;不对称肌肉受累是其特征性的MRI表现。
4)肌肉组织病理检查:
肌肉病理呈肌营养不良样或肌病样病理改变,表现为肌纤维大小不等、结缔组织增生、坏死、再生、核内移;部分患者可在肌内衣或血管周围见到炎症细胞浸润。肌肉病理改变无特异性,但可用于排除其他疾病。
5)其他检查:
肺功能需作为常规检查项目,特别是有严重躯干和骨盆带肌无力或坐轮椅的患者需要定期监测呼吸功能。对于早发型FSHD患者,建议进行眼底镜检查或荧光素眼底血管造影检查以排除Coats综合征。另外,还需定期进行听力筛查。
6)基因检测:
详见本文“分子遗传诊断与分型”。
FSHD目前尚无特异性的治疗手段,主要是对症治疗缓解相关症状。对于存在疼痛的患者,可口服非甾体抗炎药缓解患者的急性疼痛,抗抑郁药及抗癫痫药可缓解患者的慢性疼痛;呼吸功能障碍时,可采用辅助通气技术;腰椎前凸严重影响患者站立、坐立体位时,可以考虑借助一些支撑设备,如腹带、腰托等;可通过肩胛固定术提高肩关节的功能;足下垂可以通过穿戴足托来矫正;患者日常需防止跌倒。此外,康复治疗是FSHD的重要治疗方式,低强度的有氧运动(步行、游泳及做韵律操等)可提高患者的肌力、改善患者生活质量。
降低DUX4基因的表达或DUX4蛋白的活性是治疗FSHD的重要靶点,如通过反义寡核苷酸或干扰RNA技术抑制DUX4mRNA的转录水平,达到改善肌肉损害的目的,为FSHD的治疗带来了曙光。但是目前靶向治疗的疗效尚不明确,需要进一步的临床证据。通过抗氧化剂减缓疾病的进程,也可能是FSHD的治疗方式。
遗传咨询和产前诊断是预防本病的关键。对于有家族史的孕妇,可在妊娠早期或中期取绒毛细胞或羊水细胞抽提DNA,然后应用Southern杂交方法进行产前诊断。产前诊断主要包括:
1.70%~90%先证者的父母之一有D4Z4重复单元的减少,10%~30%先证者由新发突变所致;当先证者家庭成员表现为“阴性”家族史时,需考虑其父母可能带有D4Z4重复单元突变而处于症状前状态或在发病前已去世。
2.先证者同胞的患病风险取决于其父母的遗传学特征,如果父母之一带有突变的基因,先证者的同胞有50%的可能性患病;如果先证者的双亲在外周血细胞DNA中都没有检出该突变基因,可能是父母任何一方的性腺镶嵌体。在这种情况下,同胞患病的风险增高。先证者的子女有50%可能遗传D4Z4重复单元缩短这一变异。
3.考虑到该病的外显率在不同年龄和性别之间有显著差异,对先证者家族成员进行家系分析和DNA诊断的检测,有可能发现症状前阳性个体,故须慎重实施这种症状前诊断,因目前无有效的治疗方法,会给症状前患者的心理及工作带来不良的影响。
在确定了父或母的D4Z4重复单元减少的基础上,可采用Southern印迹杂交法进行高风险胎儿的产前诊断。









