


 收藏
收藏 已收藏
已收藏英文名称 :spondyloepiphyseal dysplasia
脊椎骨骺发育不良(spondyloepiphyseal dysplasia,SED)为一组选择性累及脊柱和长管状骨骨骺的染色体遗传性发育障碍性疾病。根据《国际遗传性骨病分类标准(2015年版)》分为先天性、迟发性和其他罕见类型,如Kimberley型、Wolcott-Rallison型等。临床上有躯干与肢体不成比例矮小的特征。发病率低,为(0.1~0.4)/100 000,是一种较少见的骨软骨发育不良。脊椎骨骺发育不良的软骨细胞病理学特点是关节软骨细胞核增大,大量蛋白多糖沉积,核染色质聚集,粗面内质网明显扩张。软骨细胞成熟障碍,胶原和蛋白多糖在内的细胞外基质分泌障碍可能是本病关键性病理特点。
先天性脊椎骨骺发育不良为常染色体显性遗传病,发病率约为1/100 000。迟发性脊椎骨骺发育不良为X-连锁隐性遗传疾病,实际发病率尚未确定,在英国人口中统计约为1.7/1 000 000。
COL2A1基因的杂合突变是SEDC最常见的病因。大多数SEDC的发生通常与12号染色体长臂上编码Ⅱ型胶原的COL2A1基因突变有关。COL2A1基因的杂合子突变导致的一组疾病总称为Ⅱ型胶原病(MIM 120140)常见的临床表型包括Ⅰ型Stickler综合征(MIM 108300)、Kniest发育不良(MIM 156550)、Ⅱ型软骨成长不全(MIM 200610)、骨关节病(MIM 165720)、Wagner综合征(MIM 143200)、Strudwick型脊柱骨骺干骺端发育不良(MIM 184250)、脊柱干骺端发育不良(spondylometaphyseal dysplasia,SMD,MIM 184252)和不常见的表型,包括双侧缺血性股骨头坏死(MIM 608805)、脊柱周围发育不良(spondyloepiphyseal dysplasia,SPPD,MIM 271700)、Torrance型扁平椎体致死型骨骼发育不良(platyspondyliclethal skeletal dysplasia,Torrance type,PLSDT,MIM 151210)。
COL2A1基因编码Ⅱ型胶原α1链,3条单一的前α1链构成Ⅱ型胶原蛋白。Ⅱ型胶原蛋白是在Ⅱ型前胶原蛋白的基础上加工成熟而成的。Ⅱ型胶原蛋白由3条单一的前α1链构成。前α1链中间含有300个Gly-X-Y重复序列,Gly为甘氨酸,X位置上常常是脯氨酸,Y位置上常常是羟基脯氨酸残基,在Gly-X-Y重复序列的两侧是N端和C端前肽。前α1链首先经过转录后的修饰,包括Y位置的脯氨酸残基的羟化,然后折叠成成熟稳定的三螺旋前胶原蛋白分子分泌到细胞外。经酶裂解N端和C端前肽,使Ⅱ型前胶原蛋白成熟为Ⅱ型胶原蛋白。在纤维形成的早期阶段,胶原分子相互作用的关键依赖于胶原分子特异的Gly-X-Y重复序列的位点。另外,Ⅱ型胶原纤维的形成还受其他大分子如蛋白聚糖和非纤维的胶原的调节。因此,COL2A1基因的突变,导致了Ⅱ型胶原病的发生。
SEDL基因在进化中高度保守,420bp的开放阅读框编码含140个氨基酸的蛋白质Sedlin。Sedlin蛋白功能尚不清楚,对sedlin蛋白功能的研究发现:sedlin亚细胞定位与核周高尔基复合体分布一致,而发生移码突变失活的SEDL基因产物在细胞内分布异常;对sedlin的cDNA进行序列分析发现其编码产物的羧基端存在潜在的内质网滞留信号;酵母同源蛋白P20参与构成转运蛋白粒子复合体,其在内质网至高尔基复合体的运输囊泡与受体之间的定位和融合过程中起关键作用。另外,发现sedlin羧基端序列可能具有与高尔基复合体相互作用的特征。据这些现象推测,sedlin可能参与细胞内蛋白质等从内质网到高尔基之间以囊泡形式运输的过程。但其具体的作用机制及其在软骨内骨生成过程中所起的作用尚未完全明确。
根据发病时间分为先天性脊柱骨骺发育不良(spondyloepiphyseal dysplasia congenita,SEDC,MIM 183900)和迟发性脊柱骨骺发育不良(spondyloepiphyseal dysplasia tarda,SEDT)。后者以X-连锁迟发性脊椎骨骺发育不良(X-linked spondyloepiphyseal dysplasia tarda,SEDL,MIM 313400)最为多见。
1.生化检查
血钙、磷和碱性磷酸酶正常,血尿生化无改变。合并骨质疏松时,PTH水平升高。
2.X线检查
先天性脊柱骨骺发育不良:新生儿期X线改变不明显,仅可见脊柱、骨盆、四肢骨化延迟。随年龄增长表现出椎体不规则骨化,发育不良,形成严重的扁平椎及脊柱后凸畸形。齿状突亦常发育不良,髂骨翼变短、变方,体部相对宽大,髋臼呈水平位且加深。耻骨化骨棱延迟、骨化差。股骨头、颈发育不良,髋内翻。胸廓前凸呈铃形。
X-连锁迟发性脊椎骨骺发育不良(图1):脊椎椎体普遍变扁、呈舌样变,椎体后部呈驼峰样凸起,椎间隙中后部变窄,以胸腰段明显。椎弓根发育正常,椎弓间距及椎管矢状径无明显改变。椎体小关节模糊、椎体韧带部分骨化。骨盆发育较小,髂骨翼变小、变方,闭孔径髋臼角加大。股骨头、颈发育较小。髋、膝、踝、肘、腕等关节较早出现退变,骨质疏松明显。头颅骨及手掌指骨均无明显改变。
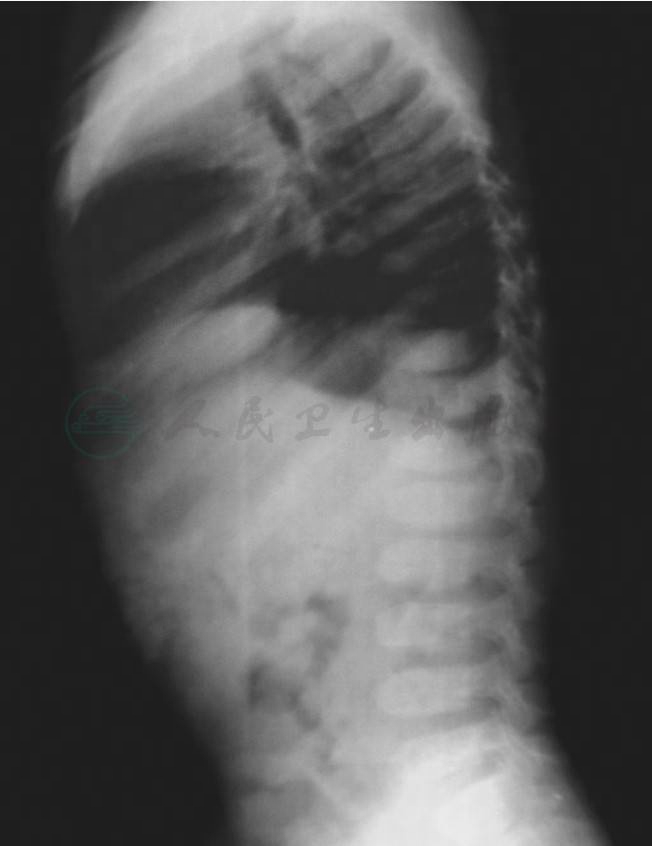
图1X-连锁迟发性脊椎骨骺发育不良脊柱X线片
椎体普遍变扁、呈舌样变,椎体后部呈驼峰样凸起,椎间隙中后部变窄
此类疾病目前尚无有效的治疗方法。严重者在生后因缺氧等死亡,成年后缓解关节疼痛,可能需骨骼矫正或关节置换术,治疗腭裂、近视等并发症;晚发型患儿成年后应尽量避免可能损伤脊柱和大关节的运动,可延缓其进展。积极治疗并发症,最常见的是髋关节发育不良,可能需要进行髋关节置换术;还可进行脊柱校正。同时应预防早期关节炎的出现,并且适当进行心理治疗和开展遗传咨询。有关数据表明:生长激素的使用无效果。
进行产前检查,保证优生是预防此类疾病的有效方法,降低疾病的发生率。









