


 收藏
收藏 已收藏
已收藏英文名称 :congenital malformations of the anus and rectum
先天性肛门直肠畸形(congenital malformations of the anus and rectum)占消化道畸形第一位,世界范围内发病率为1/1500~1/5000,据国内文献报道我国的发病率为1/2800。男、女发病率大致相等,男性稍多。先天性直肠肛门畸形不仅发病率高,而且种类繁多。肛门直肠畸形病理改变复杂,不仅肛门直肠本身发育缺陷,肛门周围、盆底肌肉及神经也均有不同程度的改变。此外,该畸形常合并有其他器官的畸形。
在胚胎第三周末,后肠末端膨大与前面的尿囊相交通,形成泄殖腔(cloaca)。第四周位于泄殖腔与后肠间的中胚层皱襞形成并向尾侧生长,同时间充质于泄殖腔二侧壁的内方增生形成皱襞称侧褶向腔内生长,构成尿直肠隔,将泄殖腔分为前后两部分,前者为尿生殖窦,后者为直肠。同时泄殖腔的尾端被外胚层的一层上皮细胞膜所封闭称为泄殖腔膜,使与体外相隔,前者为尿生殖窦膜,后者为肛膜。第七、八周时,两个膜先后破裂,肛膜破裂后肠与原肛贯通,形成正常的直肠和肛管。如后肠或原肛发育不良或贯通不全,即形成各种类型的肛门闭锁或狭窄。若尿生殖窦与后肠分隔不全后肠开口仍在尿路器官上,则形成直肠和泌尿生殖系统的瘘管或直肠会阴瘘、肛门前移等畸形。
总之,肛门直肠畸形的发生是正常胚胎发育期障碍的结果,引起的原因尚不清楚,目前认为是遗传因素和环境因素共同作用的结果。
关于肛门直肠畸形的基因研究提示,HOX、SHH、FGF基因,Gli2和Gli3转录因子可能与人类肛门直肠畸形相关。据文献报告,肛门直肠畸形有家族发生史者在1%~9%。Van Gelder报告在一个家庭中,兄弟姐妹4人均患此病,矢野博等收集的29篇文献中有34个家庭发病。对于家族发病基因的研究结果表明,肛门直肠畸形与HLA基因有关。
肛门直肠畸形也和其他畸形一样,可能与妊娠期,特别是妊娠早期(4~12周)病毒感染、化学物质、环境及营养等因素的作用有关。胚胎期发生发育障碍的时间越早,所致畸形的位置越高、越复杂。动物实验证实氯仿、乙烯硫脲、全反式维A酸、阿霉素等均可诱导母鼠产生肛门直肠畸形鼠仔,说明这些药物是使妊娠动物产生肛门直肠畸形的直接原因。
1970年制定的国际分类,以直肠末端与肛提肌,特别是与耻骨直肠肌(Braune’s muscle)的关系为标准,将肛门直肠畸形分为高位、中间位和低位三型,直肠盲端终止于耻骨直肠肌环以上者为高位畸形,位于该肌之中并被其包绕者为中间位畸形,穿过该肌以下者为低位畸形。每型又分有瘘和无瘘两组,瘘的发生率约占50%,尤以女孩为多。女孩有直肠阴道瘘、前庭瘘及会阴瘘,男孩有直肠膀胱瘘、尿道瘘及会阴瘘(图1~3)。
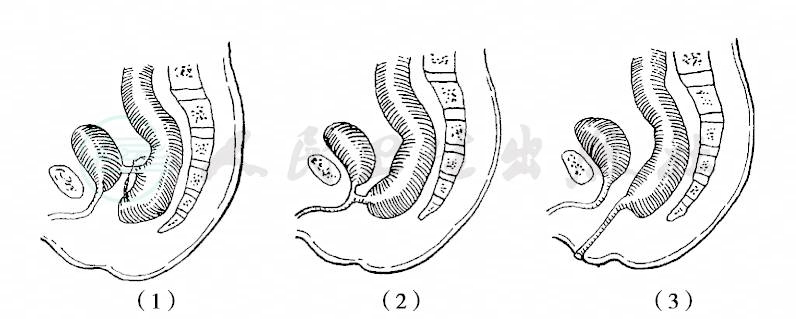
图1 男孩无肛合并瘘管
(1)直肠膀胱瘘;(2)直肠尿道瘘;(3)直肠会阴瘘
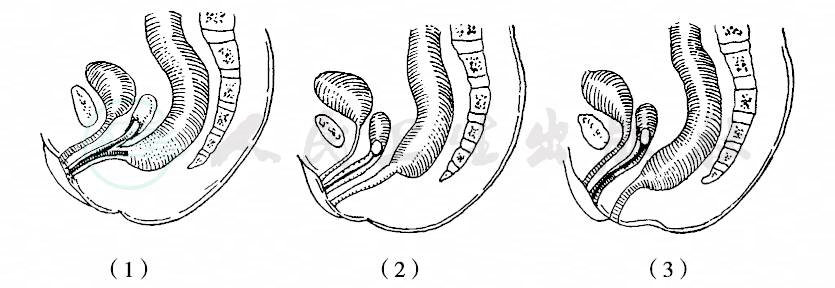
图2 女孩无肛合并瘘管
(1)直肠阴道瘘;(1)直肠前庭瘘;(3)直肠会阴瘘
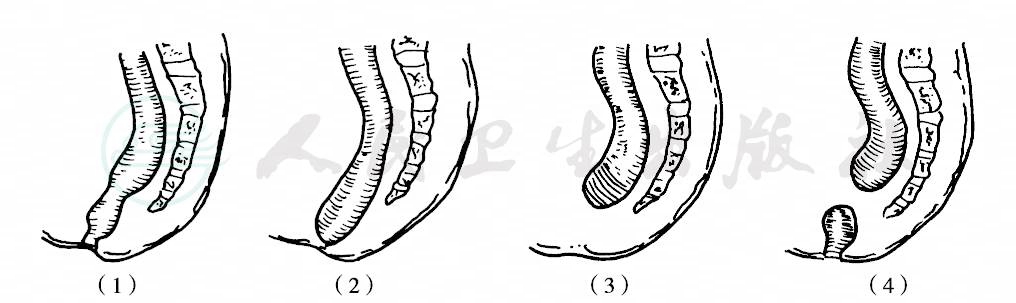
图3 无瘘肛门直肠畸形
(1)肛门狭窄;(2)低位肛门闭锁;(3)高位闭锁;(4)肛管正常直肠远端闭锁
1984年,Wingspread将分类法加以简化,具体分类见表1。
表1 先天性直肠肛门畸形的分类

近年来,对先天性肛门直肠畸形患儿盆底组织结构研究的结果表明,盆底肌肉、骶骨、神经及肛周皮肤等均有异常,畸形位置越高,病理改变程度越重。
1.肌肉改变
肛门直肠畸形患儿的肛提肌,包括耻骨直肠肌的发育良好,仅个别病例该肌缺如或发育不良。由于畸形类型不同,耻骨直肠肌的位置可发生改变,高位畸形该肌明显向上向前移位,并短缩,呈闭锁状,依附于前列腺、尿道或阴道后方,并与直肠盲端和外括约肌有一定距离。中位畸形时直肠盲端位于耻骨直肠肌中。直肠前庭瘘(rectovestibular fistula)和低位畸形耻骨直肠肌基本位置正常。
镜下观察外括约肌走形方向,在正常儿外括约肌肌纤维呈横断面,低位畸形肌纤维也呈横断面,中位畸形外括约肌肌纤维排列呈斜行,高位畸形外括约肌发育不良,肌纤维走行方向紊乱,或仅有痕迹。
内括约肌在高位畸形缺如,中位畸形发育差,低位畸形内外括约肌发育正常。
2.神经改变
肛门直肠畸形患儿常伴有骶椎畸形(sacral vertebral deformities),当骶椎椎体缺如时,可伴有骶神经的改变,缺如的节段越多,骶神经改变越明显,可直接影响该病的治疗与预后。
3.伴发畸形
肛门直肠畸形常合并其他先天性畸形,其发病率占40%~70%,最常见者为泌尿生殖系畸形,其次为脊柱,特别是骶椎畸形,再次为消化道、心脏以及其他各种畸形。肛门直肠畸形可以伴发几种畸形,例如肛门闭锁合并骶椎畸形、骶前肿物称Currarino综合征。这些伴发畸形增加了治疗上的困难,并可影响预后。因此,对肛门直肠畸形患儿应进行全面的检查。
先天性肛门直肠畸形的治疗方法,根据其类型及末端的高度而不同。凡无排便功能障碍如会阴前肛门无狭窄者,无需手术治疗外,其他先天性肛门直肠畸形均需手术治疗。
低位无肛如膜状闭锁、狭窄或直肠会阴瘘均需会阴部肛门成形术。
中、高位无肛如直肠尿道瘘、直肠膀胱瘘及直肠阴道瘘等可行后矢状入路肛门成形术(Pena)或腹腔镜辅助肛门成形术(laparoscopic assisted anorectoplasty)。
泄殖腔畸形(cloacal deformity)的患儿出生后应立即行结肠造瘘,使分流改道,保持泄殖腔清洁,防止尿路感染。根治手术的时间应根据患儿情况、畸形复杂程度而定,以6个月后为宜。手术可取正中矢状切口,充分游离泄殖腔管,显露直肠进入泄殖腔的入口,于直肠和阴道共壁之间做黏膜下分离,分离直肠的长度直至能无张到达肛门皮肤为止。同样的方法分离共壁的阴道与尿道,成形阴道与尿道,若阴道不能到达会阴部皮肤,可采用皮肤阴道成形术或肠管阴道成形术。
为了防止肛门成形术后瘢痕狭窄,均应于术后2周进行扩肛,持续6个月左右。泄殖腔畸形术后在扩肛的同时,需扩张阴道,阴道扩张需持续到青春期。









