


 收藏
收藏 已收藏
已收藏英文名称 :porokeratosis
汗孔角化症(porokeratosis,PK)是一组遗传性角化异常性皮肤病。Vittorio Mibelli于1893年首次报道了一例男性患者,2岁开始在手臂等处出现大小不一、中央萎缩、边缘呈堤状隆起的环型皮损,Mibelli认为该病是由汗管空细胞过度角化所致,故将其命名为“汗孔角化症”,该病例为经典的斑块型汗孔角化症(porokeratosis of mibelli,PM)。1937年,Andrews首次报道了播散性浅表性汗孔角化症(disseminated superficial form of porokeratosis,DSP)。1969年,Chenosky等发现了光化性浅表播散型汗孔角化症(disseminated superficial actinic porokeratosis,DSAP),该型皮损与DSP相似,但主要发生于光暴露部位。1971年,Guss等首先提出了掌跖播散型汗孔角化症(porokeratosis palmaris et plantaris disseminata,PPPD),该型皮损首先侵及掌跖,逐渐泛发全身,包括光暴露部位和非光暴露部位。1971年,Goldner R首次报道了线状型汗孔角化症(linear porokeratosis,LP),该型皮损沿Blaschko线呈簇状或线状排列于肢体或躯干,面部受累少见。1977年,RRahbari等首次提出点状型汗孔角化症(porokeratosis punctata,PP),此型多于成年发病,皮损为局限于掌跖部位的粟粒样角化斑点,边缘稍隆起,触之柔软。
除上述经典分型外,还有一些罕见的特殊类型,如网状PK、脓疱型PK、瘙痒性发疹性丘疹型PK、皱褶部位反转型(疣状)PK、局限于生殖器的PK、溃疡型PK、大疱型PK、结节性痒疹样PK、日光性面部PK、汗孔角化棘皮瘤、脂溢性角化样PK、面部毛囊PK等。上述类型中,一部分仅为病例报道,一部分日渐为学者所熟知。
汗孔角化症是一组遗传性角化异常性皮肤病,多数PK表现为常染色体显性遗传,但也有部分为散发病例。除遗传因素外,紫外线、感染、外伤和免疫抑制等因素也可能参与疾病发生。
(一)遗传因素
目前所报道的家族遗传性汗孔角化症病例均为常染色体显性遗传模式,伴不完全外显率。而局灶性汗孔角化症可能通过镶嵌现象发病,即体细胞突变导致的局灶性杂合性缺失。遗传所致或者散发的基因缺陷导致免疫功能及角质形成细胞功能变化,引发鸡眼样板形成,而此结构是本病的主要病理学特征。
目前已知MVK、PMVK、MVD、FDPS基因突变均可能参与PK的发病,其中DSAP/DSP致病基因多为MVD(58%),亦包括MVK(27%)、FDPS(4%)基因,LP致病基因多为MVD(30%),同时可见PMVK(10%)、MVK(10%)基因,有的病例并未检测出突变基因。
(二)紫外线
有研究显示,汗孔角化症皮损暴露于日光之后瘙痒感会加重,而在冬季,瘙痒症状将会减轻。曾有研究指出,紫外线可能会影响所照射区域皮肤的免疫功能,故应避免紫外线照射。
(三)感染性因素
曾有学者将PK的皮损标本注入豚鼠腹部皮下,造成豚鼠感染并发生类似PK的表皮改变。另有报道发现,合并多系统感染的患者出现了PK的临床表现,故认为感染可能是PK的一种致病因素。
(四)外伤
有报道发现,利用外科手术切除PK皮损后,在创面处出现类似PK的临床表现,另也有报道发现在烧伤部位出现PK皮损,这些均表明皮肤损伤可能是诱导PK发生的因素之一。
(五)免疫抑制
免疫抑制诱导PK的发生近年来多有报道。Schamroth研究发现,服用泼尼松、环磷酰胺等免疫抑制药物后,在大腿、手臂等部位发生弥漫性汗孔角化症,发病时间1周到16年不等,提示免疫抑制可能会诱导汗孔角化症发生。
汗孔角化症临床表现多种多样,为避免分类上的混乱,Schamorth在1997年将汗孔角化症分为3大临床类型:局限性、播散性和免疫抑制诱导性。局限性PK包括PM、LP和PP;播散性PK包括DSP、DSAP和PPPD。近些年有报道发现,接受了器官移植、免疫抑制剂治疗及合并HIV感染的患者更容易发生汗孔角化症,故而提出免疫抑制诱导性PK这一相对较新的概念。
(一)斑块型汗孔角化症
经典的Mibelli汗孔角化症较为罕见,可为常染色体显性家族遗传,但更多表现为散发病例。PM常于婴儿或儿童期发病,男女比例约为2.17∶1,皮损初发为漏斗状角质丘疹,后逐渐扩展为环状、不规则斑块状,直径几毫米至几厘米不等。斑块边缘角化隆起呈嵴状,中央皮肤常萎缩。好发于四肢,以手、足居多,面、颈、肩、生殖器及其他部位均可受累,通常无自觉症状,病程缓慢,皮损可持续多年不变化。1942年,Vigne首次报道了PM的恶性转归,并指出7%~11.6%的PM患者皮损可能发生癌变,其中皮肤鳞状细胞癌最为常见。
(二)播散性浅表性汗孔角化症
该型部分病例符合常染色体显性遗传,多在10~30岁发病。皮损初起为红色斑疹或色素沉着角化斑,中间萎缩,逐渐扩展成表浅的环形(图1)。其发病与光照无关,故光暴露部位和非光暴露部位均可受累,常见于躯干、生殖器及掌跖部位。瘙痒是DSP的一个特征,约1/3的患者自觉皮损处有瘙痒感。

图1 播散性浅表性汗孔角化症
主要表现为前胸散在绿豆至蚕豆大小暗褐色环形、类圆形边缘隆起性斑片
(三)光化性浅表播散型汗孔角化症
DSAP为汗孔角化症中最常见的类型,符合常染色体显性遗传。一般青春期发病,30~40岁表型完全外显,男女比例约为1.76∶1。临床表现与DSP相似,常为数个至数百个不等的直径1cm的环状皮损,可融合成多环状,皮损处往往乏汗,少数患者自觉有瘙痒感(图2)。皮损好发于日光暴露部位,如下肢、前臂、上臂、肩背部伸侧,面颊部较少受累(约见于15%病例),紫外线可诱发和加重皮损,约50%的患者经夏季日光或人工UVA/UVB照射后皮损会加重,在冬季皮疹可逐渐消退。
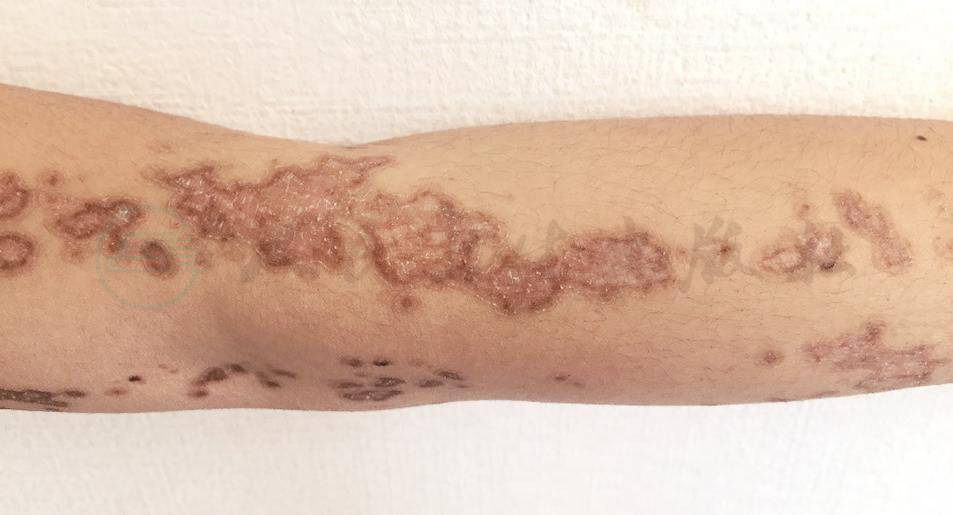
图2 光化性浅表播散型汗孔角化症
主要表现为左臂部条带状分布暗褐色环状斑,部分融合成片
(四)掌跖播散型汗孔角化症
一般在20岁前发病,男女发病比例接近2∶1。皮损初发为多个红色或棕色浅表丘疹,中央含有角栓,限于掌跖部位。几个月后围绕中心扩大,直径约4~5cm。皮疹逐渐播散至全身,包括四肢及躯干,偶有黏膜受累。
(五)线状型汗孔角化症
多在婴儿或儿童时期发病,偶见于成人,极少数为先天性,男女发病均等。皮损为单个、间断、环形的角化性丘疹或斑块,棕色或棕红色,呈线性排列,好发于肢端,不限于光暴露部位。不因阳光照射而加重。
(六)点状型汗孔角化症
多在成年后发病,男性多于女性,皮损为多发针尖或刺状角化型丘疹,边缘稍隆起,触之柔软,可融合成斑块状,直径0.5~1.5cm不等,皮损限于掌跖部位,不向其他部位发展。
汗孔角化症有恶变风险,多发生在线状型,且多见于下肢。最常见为鳞状细胞癌,其次为Bowen病和基底细胞癌。
汗孔角化症的治疗方法种类较多,治疗效果差异较大,且均不能防止复发。目前,本病在治疗方面仍缺乏大样本循证医学研究,大部分治疗仅为案例报道。对大多数患者而言,避免日光照射、局部应用保湿剂是必须的,此外应注意定期随访,警惕皮损恶变的风险。
(一)外用药物
外用10%水杨酸软膏或0.05%~0.1%维A酸乳膏可使部分PK皮损消退,但停药后常复发;5-氟尿嘧啶(5%)可用于PM、LP、DSP及DSAP患者,局部封包治疗可提高疗效。有报道一位PK患者在应用5-氟尿嘧啶联合17%的水杨酸封包治疗13个月后,皮损被成功治愈;5%咪喹莫特乳膏可用于PM、LP及疣状汗孔角化症的治疗。有报道应用5%咪喹莫特治疗疣状汗孔角化症,每周3次,应用8周后皮损消退,随访一年皮损并无复发;一些DSAP患者应用维生素D3衍生物如卡泊三醇、钙泊三醇治疗可取得较好疗效;外用糖皮质激素可使病情得到暂时缓解,但其免疫抑制作用有促进PK皮疹扩散及恶性转化的风险,故用药需谨慎;此外,有报道外用3%双氯芬酸凝胶治疗DSAP及生殖器部位PK患者,每天2次,持续3~6个月取得了较好疗效。
应用甲基氨基酮戊酸光动力疗法成功治疗DSAP的病例近些年也多有报道。但有系统性回顾文献指出,光动力疗法用于治疗DSPA患者成功率较低,治疗效果较差,副作用也较其他药物治疗更多见。
(二)系统用药
口服维A酸类药物,如依曲替酯、异维A酸、阿维A治疗PK可取得较好疗效,但此类药物的副作用限制了其长期应用。有报道发现,存在免疫抑制的PK患者口服维A酸类药物可降低皮损癌变的风险,但患者停药后皮损也多有复发。
(三)其他治疗
当PK皮损数目较少时可选用液氮冷冻、CO2激光、585nm脉冲染料激光、Nd:YAG激光、电解术、刮除术、磨削术、超声引导下外科吸引术等方法治疗,部分患者可获得较好疗效。目前无研究发现预防性的非手术性治疗能降低皮损恶变的概率,发生恶变的皮损应尽早手术切除,有报道指出显微外科手术能更加精确地将肿瘤组织从汗孔角化症皮损组织中区分开来。









