


 收藏
收藏 已收藏
已收藏英文名称 :cyst of the liver
肝囊肿(cyst of the liver)是较常见的良性疾病,分寄生虫性和非寄生虫性两类。前者以肝包虫病为多见,后者可分为先天性、创伤性、炎症性和肿瘤性囊肿。通常所称的肝囊肿是指先天性肝囊肿,其又可分为单发性和多发性两种。单发性肝囊肿较少见,小者囊液仅数毫升,囊肿大者囊液可达10000ml以上。肝内有两个以上囊肿者即为多发性肝囊肿;左右半肝有散在的大小不等囊肿又称多囊肝,这种病例大多合并多囊肾,也可同时合并胰腺、脾、卵巢、肺等囊肿。
大多数非寄生性肝囊肿不论单发或多发均为先天性。先天性肝囊肿的成因是遗传性疾病,指亲代的生殖细胞或受精卵的遗传物质发生突变而导致后代表型异常的一类疾病。表型异常可以是宏观的躯体畸形,也可以是代谢过程的异常。遗传性肝脏病亦不少见,它是指在遗传因素基础上,又由不同程度的环境因素作用所形成的,表现为肝脏形态结构和功能异常,也可伴有肝外系统损害;少数因酶的结构基因发生突变而致酶蛋白量和活性的变化,从而引起代谢紊乱。
先天性肝囊肿可能系肝内小胆管或毛细胆管发育过程中形成肓端,未与肝管分支相接,肝小叶分泌的胆汁积聚,形成囊肿。1957年Dulgaad在对284例双侧多囊肾病例及其亲属的研究中发现,173例尸检中有64例合并多发性肝囊肿,而多囊肾病例中70%有家族史,因而指出这两种先天性病变是由单基因发生的。多囊肝可发生于同一家族的不同成员,因而多囊肝的发生可能与染色体阳性或阴性遗传有关。
与多囊肝病明确相关的基因已发现有4个:分别为多囊肾基因PKD1和PKD2及独立型多囊肝基因PRKCSH和SEC63,下面分别介绍这些基因及相关蛋白。
1.PKD1和多囊蛋白‐1
1985年,通过基因连锁分析技术,多囊肾相关基因PKD1被定位于16p12.3‐p13.12。PKD1基因的突变与大约85%的ADPKD发病有关。PKD1基因的突变超过230种,均分布于整个基因,主要是错义和无义突变,亦报道有涉及mRNA拼接和基因重排的突变。PKD1编码14.1‐kb信息PKD1转译座物为多囊蛋白‐1,含4301个氨基酸,为一种膜蛋白,多囊蛋白‐1和多囊蛋白‐2在细胞膜上形成复合物,共同调节钙通道和细胞信号传导。一些研究结果提示某些类型或位点的突变可以预示疾病的表现型,如5端的突变将预示着病程进展更快,终末期肾病更严重。
2.PKD2和多囊蛋白‐2
1993年,第二个多囊肾相关基因PKD2被定位于4q21‐q23。PKD2编码的多囊蛋白‐2含968个氨基酸,分子量约110kD,亦是一种膜蛋白。已发现的PKD2突变超过60种,亦均分布整个基因。但到目前为止,尚未能确定某一突变‐5某表现型明确相关。PKD1突变为ADPKD家族中基因突变的85%~90%,PKD2则占5%~10%。生物物理研究证明多囊蛋白‐2起着阳离子通道的作用。
PKD1多囊蛋白和PKD2多囊蛋白形成复合体位于肾初级纤毛、胆管上皮细胞初级纤毛,能感知流过肾小管的液流以及将液流引起的机械刺激转化成为钙离子信号向细胞内传递,调节胆管和肾上皮细胞液体分泌,及生长和增殖。正常情况下液流跨越细胞表面弯入初级纤毛,激活钙离子流入细胞,亦抑制液体分泌。PC‐1和PC‐2复合体的作用已被针对PC‐2的通道阻断抗体或干扰PC‐1合成的基因突变所证实。据推测,多囊蛋白‐1和‐2组成了一个信号传导系统,通过改变G蛋白的信号传导过程和调节细胞内钙离子水平,将细胞刺激转化成细胞内信息,且多囊蛋白‐2介导的钙离子内流可为钙离子活化型钙离子通道进一步增强,ADPKD患者囊上皮细胞表达癌基因和生长因子受体,提示PC‐1和PC‐2在核内调节作用。PC‐1调节mTOR活性,而mTOR在蛋白翻译、细胞生长和增殖中起着重要作用。研究提示,PC‐1突变或纤毛损坏可导致纤毛内PC1/mTOR抑制复合物丢失,进而引起细胞增生和囊肿形成。
3.PRKCSH基因与肝囊肿蛋白
临床上发现多囊肝独立于多囊肾而单独发病,基因上与PKD1,PKD2无关。2000年通过基因连锁分析,一个独立型多囊肿相关的基因被定位于19p13.2‐13.1,为一多功能基因序列,但基于已知多囊蛋白1或2的结构或功能并未发现相似的功能基因。后续研究表明产生突变的是PRKCSH基因。PRKCSH基因编码的蛋白为一已知蛋白,即蛋白激酶C底物80K‐H或葡萄糖苷酶Ⅱβ亚体,鉴于其与多囊肝相关,对应于多囊蛋白1和2,称为肝囊肿蛋白(hepatocystin)。肝囊肿蛋白由527个氨基酸残基组成,分子量约59KD。已报道肝囊肿蛋白功能,对葡萄糖苷酶Ⅱ蛋白复合物的非催化亚单位起初级作用,修饰蛋白糖基化和后转录程序合成新糖蛋白。多囊肝表现型可能是由于肝囊肿蛋白正常功能丢失而引起。免疫痕迹研究亦证明,多囊肝患者中肝脏全长囊肿蛋白表达量明显降低,不足正常肝组织表达量的一半,而多囊肝囊肿壁中正常肝囊肿蛋白表达水平更低,只达正常肝组织的13%。
4.Sec63和Sec63蛋白
2001年发现了第二个独立型多囊肝基因—SEC63基因,位于6号染色体,含21个外显子,突变部位位于外显子2~19之间。PCLD与内质网和蛋白程序有关功能被Sec63基因增强,Sec63基因编码蛋白命名为Sec63蛋白,是内质网内结合膜蛋白,其功能作为蛋白转送复合物组成部分,此蛋白程序步骤是葡萄糖苷酶Ⅱ执行的糖基化修饰步骤的上游。
Sec63可能存在两个致使途径:翻译协同途径(信号识别体依赖性途径)和翻译后途径(非信号识别体依赖途径),在哺乳动物中,翻译协同是主要途径,核糖体直接与Sec63转录子联合并及将新合成的多肽挤出核糖体。
多囊性疾病之间分子联系可能是Sec63,GⅡβ及多囊蛋白1、2和纤维囊肿蛋白作用的对象是同一种蛋白。
非寄生虫性肝囊肿由Briston在1856年首先报道,认为属少见病,1955年Melnic报道687例尸检仅发现一例。上海医科大学附属中山医院及上海第二医科大学附属瑞金医院统计4448例尸检中,先天性肝囊肿的检出率为0.22%。林芷英根据尸检和剖腹手术资料,其发病率为0.14%~0.53%。梁扩寰等于1983—1988年共行肝脏声像图检查20980例,发现肝囊肿213例,检出率为1.02%。湖南省人民医院肝胆外科门诊患者中肝囊肿发病率为1.6%。多发性肝囊肿比单发性多见,约半数患者同时合并肾、胰、脾、肺、脑或卵巢等囊肿。常侵犯左、右半肝亦有局限于肝一叶。吴孟超报道PCLD比单发性肝囊肿多见,尸检检出率约0.15%~0.5%,本病多见于40~60岁女性。
国外报道PCLD确切发病率不明,尸检发病率为0.05%~0.13%。有常染色体显性遗传多囊肿肾病(ADPKD)的患者,多发性肝囊肿发病率45%~68%,ADPKD的囊肿病变发病率大约从30余岁的24%增至60余岁时的80%。ADPKD与PCLD并存的患者占16%~93%,此范围很大,因为ADPKD时的PCLD发病与患者年龄和肾功能异常程度有关。
当ADPKD发生时,从青春期开始肝囊肿开始形成,尽管肝囊肿与ADPKD的男女比例相同,但女性趋于发生更多和更大的囊肿。囊肿形成被认为雌激素有关,因为其数目和大小与妊娠次数、服用避孕药及使用女性激素替代疗法有关。临床多囊性疾病与年龄增加、囊性肾病的严重性及肾衰竭有关。
大部分作者认为PCLD主要是无症状性疾病,由于血液透析和肾移植的成功会相继出现症状,有肾功能不全的老年患者更多地会有更多和更大的肝囊肿。有报道表明,进行血液透析的ADPKD患者中有10%的病死率可能与PCLD有关。
以前的尸检结果和外科认为,在所有多发肝囊肿患者中,50%的患者无肾囊肿,这似乎估计过高。最近的尸检结果表明只有7%的PCLD患者无肾囊肿。在患者的宗亲中有多人的肝内有多发囊肿,但无ADPKD。
1.与肝囊肿病进展有关的因子
ADPKD临床过程有明显异质性,甚至在同胞孪生也如此,此种异质性可能部分由于部位、敏感性及PKD1和PKD2体细胞第二次打击时间有关。此外,修饰基因和其他因子也可能促进肝囊肿生长。有一些ADPKD强候选修饰基因,被证实具明显作用并成为这些候选者特征。
2.其他衍生肝囊肿生长的因子
女性激素(主要是雌性激素),腔液分泌和积聚的细胞活素(Cytokines)和生长因子都可能影响ADPKD患者中肝囊肿扩张。ADPKD和PCLD血缘关系的影像学研究,表明严重肝囊肿性病更多见于妇女,ADPKD严重肝囊性病也与妊娠次数和使用雌性激素有关,且激素替代疗法加重疾病。雌激素促发肝囊肿生长与胆管上皮细胞α和β雌激素受体表达有关。腔液分泌,上皮细胞增殖和血管形成促进囊肿增大。正常肝内胆管上皮细胞对肠激素如胰泌素起反应,人肝囊肿保留调节分泌能力,产生囊腔内正压,增加胰泌素进入静脉速率。在囊肿上皮细胞培养模型随腔内压增加上皮细胞增殖率也加快。囊肿内皮细胞传导机械力如伸张和其他类型压力,或合成、释放某种类型细胞因子和生长因子刺激细胞增殖速度。
另外,分析ADPKD患者囊液在生理上与下列因子浓度有关:IL‐8,ENA‐78,GRO‐α,VEGF和IL‐6。CXCR2是IL‐8、GRO‐α和ENA‐78的受体,能驱动细胞增殖,定位于肝囊肿上皮细胞顶端范围。有趣的是,胆管细胞和肝囊肿上皮细胞不仅释放VEGF,而且VEGF受体表达也在这些细胞。因此,自主分泌和旁分泌环建立,这些因子分泌并积聚在囊液内,结合受体和内衬上皮细胞的信号细胞增殖。在鼠研究显示VEGF对囊进展起重要作用,抑制VEGF信号延缓肝囊肿增大。
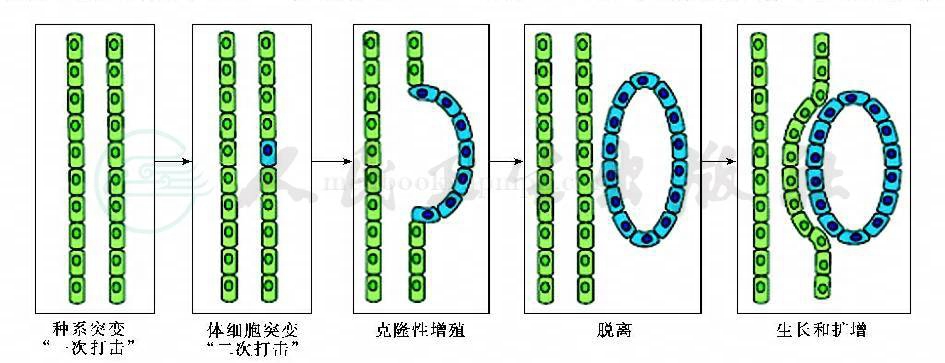
图1 常染色体显性遗传多囊性肾病的二次打击机制图
3.分子水平机制:“两次打击”学说
尽管ADPKD表现型呈常染色体显性遗传,但在分子水平上则可能呈“隐性遗传”,需要体细胞的第二次突变。“两次打击”模型(图1)要求PKD1和PKD2首先存在最初的种系突变(一次打击),接着单个体细胞受到第二次打击,相应基因拷贝发生功能缺失性突变,进而引起管腔上皮细胞的增殖,形成囊肿。在ADPKD患者身上,囊肿均在肾单位或肝内胆管树局部形成,且单个囊肿的上皮细胞均起源于单克隆,并且发病存在年龄依赖性。两次打击模型尚未在独立型多囊肝中得到证实。但亦存在这种可能,即胆道上皮细胞内囊肿蛋白功能的丢失由正常等位基因的体细胞突变引起,从而引起细胞的增殖和组织结构的变化。至于独立型多囊肝无肾脏表现可能是因为肾脏中存在蛋白成熟的替代途径,或二次突变形成纯合子引起组织特异性细胞死亡。
4.初级纤毛与多囊肾病(PKD)的发病机制
目前已有多种动物模型研究提示,初级纤毛与PKD的发病机制密切相关(图2)。PKD囊肿形成相关蛋白如多囊蛋白Ⅰ、多囊蛋白Ⅰ、polaris、polyductic和囊肿素等均先后被发现于人类和鼠类肾细胞初级纤毛中。Pazour等在IFT88蛋白突变的Tg737orpk小鼠中发现纤毛装配缺陷可导致PKD,其表现与ARPKD相似。Praetorius等证明液流刺激纤毛可引起细胞钙内流增加,且该作用依赖于纤毛结构和功能的完整性。而Nauli等进一步发现,这种液流应答需位于纤毛内的多囊蛋白参与。研究结果提示,初级纤毛的功能异常可能是形成囊肿的共同通路,其异常导致细胞—细胞和细胞—基质间的相互作用异常,引起细胞增生,细胞去分化、极性改变及促进囊液分泌,从而形成囊肿。PKD除肾脏表现外,还有肾外病理改变,如肝脏囊,胆管上皮细胞增生等。Masyuk等研究发现,胆管上皮细胞的初级纤毛也存在多囊蛋白,且纤毛也是通过机械感受器发挥功能,并进一步证实通过肝内胆管微灌注造成的管腔液流可增加细胞内钙(图3),同时降低CAMP水平。
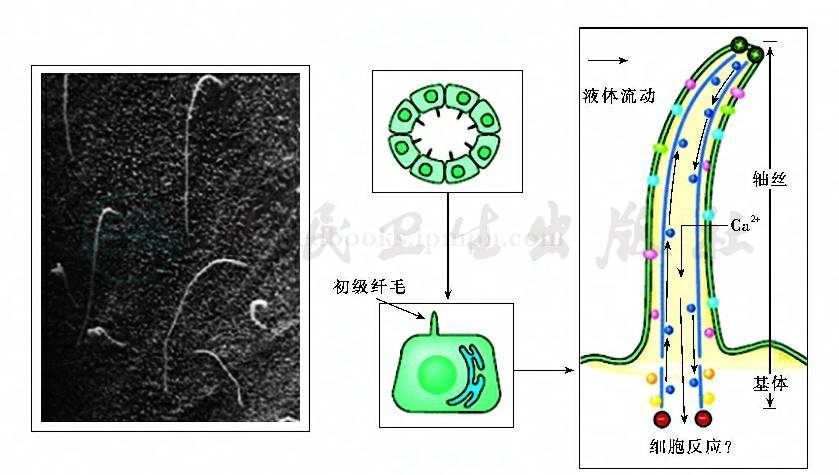
图2 初级纤毛是上皮细胞顶膜的机械感受器
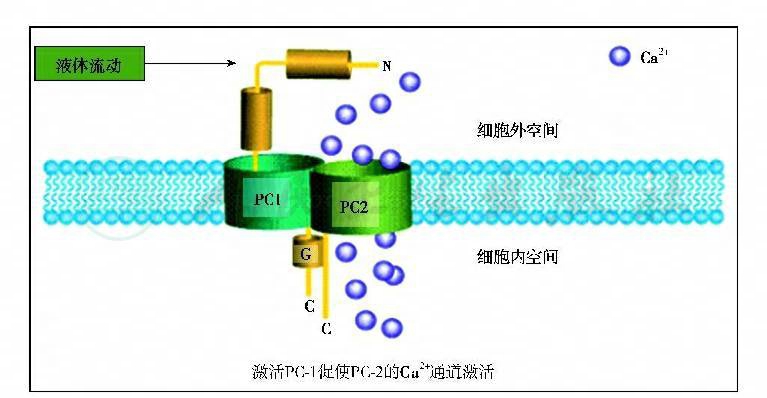
图3 PC‐1、PC‐2的相互作用及钙内流的调节
多发性肝囊肿比单发性囊肿多见,小者仅在显微镜下查到,大者容量可达1000ml以上,囊肿可散布于全肝或密集于肿的一叶,以右肝为多见。标本切面呈蜂窝状改变,囊壁可分2层,内层为上皮细胞,其形状因囊肿大小而不同。囊肿较大者,因上皮细胞受压,柱状和杯状细胞呈扁平状或消失;中等大的囊肿,只有杯状细胞;小的囊肿杯状和柱状细胞均有。外层为胶原样组织,较厚的囊壁中有较大的胆管、血管及神经。囊肿之间有较多的小胆管和正常的肝细胞,囊液澄清,一般不含胆汁。囊液为浆液性,囊液的电解质组成与血清相同,囊壁上皮可分泌多种成分,囊液的γ‐GT、CEA、CA19‐9高于血清水平,囊内压为1.5~3.92kPa(16~40cmH2O),高于胆道内压力,故囊液分泌可能属于主动分泌。
多发性肝囊肿很少引起门静脉高压和食管静脉曲张,但可合并胆管狭窄、胆管炎和肝炎。晚期可引起肝功能损害,出现腹水、黄疸、肝大、食管静脉曲张或腹壁静脉曲张。
单发性肝囊肿发生于右叶较左叶多一倍。大小不一,差别悬殊。小者仅数毫米,大者直径可达20cm,一般含液量常在500ml以上,最多可达17000ml。囊肿呈圆形或椭圆形,多为单房,亦有多房者。囊肿有完整包膜,表面呈乳白色,也有的呈蓝灰色,囊壁厚薄不一,厚者可达5mm。内层为柱状上皮细胞,外层为纤维组织,被覆有较大的胆管、血管束,周围肝细胞常受压而萎缩变性。囊液为清亮的中性或碱性液体,比重1.010~1.022,含有少量的白蛋白、黏蛋白、胆固醇、红细胞、胆红素、酪氨酸等。若合并囊内出血,可呈咖啡色。
PCLD常无特征性临床症状,确定诊断,有赖于各种影像诊断技术,超声显像诊断最具有诊断价值。诊断多发性肝囊肿的同时,还要注意肾、肺以及其他脏器有无囊肿或先天性畸形,如多囊肾,对确诊ADPKP相关PCLD很有帮助。
1.B型超声
实时声像(real‐time sonography)一般能对肝脏进行很好的检查,体瘦而合作的患者最适合进行超声检查。为了检查整个肝脏,常需要更换超声探头并将患者转至不同体位,当肝脏大部分处在右上腹部肋骨后方时尤为重要。
检查肝脏最好是用3~5MH2探头。在较魁梧的患者或当有严重的肝内疾病患者,使用一个2MH2探头可能有好处,因为较低频率探头有较高的组织穿透力。在另一方面,由于空间分辨率的提高,使用一个较高频率的探头有助于探察肝脏浅层或表面上的病变。
B超检查肝囊肿准确率达98%,而且能确定囊肿的性质、部位、大小、数目及累及肝脏的范围,为首选的检查方法。且有助于肝外腹腔囊肿的鉴别,亦可确定有多囊肾存在。
肝囊肿的声像图表现为肝内有圆形或椭圆形液性暗区,囊壁菲薄,边缘整齐、光滑,与周围组织界限清楚,囊肿后壁及深部组织回声增强,而侧壁常伴折射声影。后方回声增强产生于充有液体的结构,可以根据紧贴肿块后方的回声(发亮)区而认定。一些囊肿在囊内有回声,此情况包括囊内分隔,低度的囊内回声很可能代表沉渣及结节性囊壁。此时,该囊肿应认为是复杂性的,可能有出血或感染。虽然大多数的复杂性囊肿是良性的,结节性囊壁或患者有某种肿瘤的病史(如卵巢癌),应考虑复杂性囊肿属恶性。多发性肝囊肿在B超图像上显示为数量不一、大小不等的液性暗区。
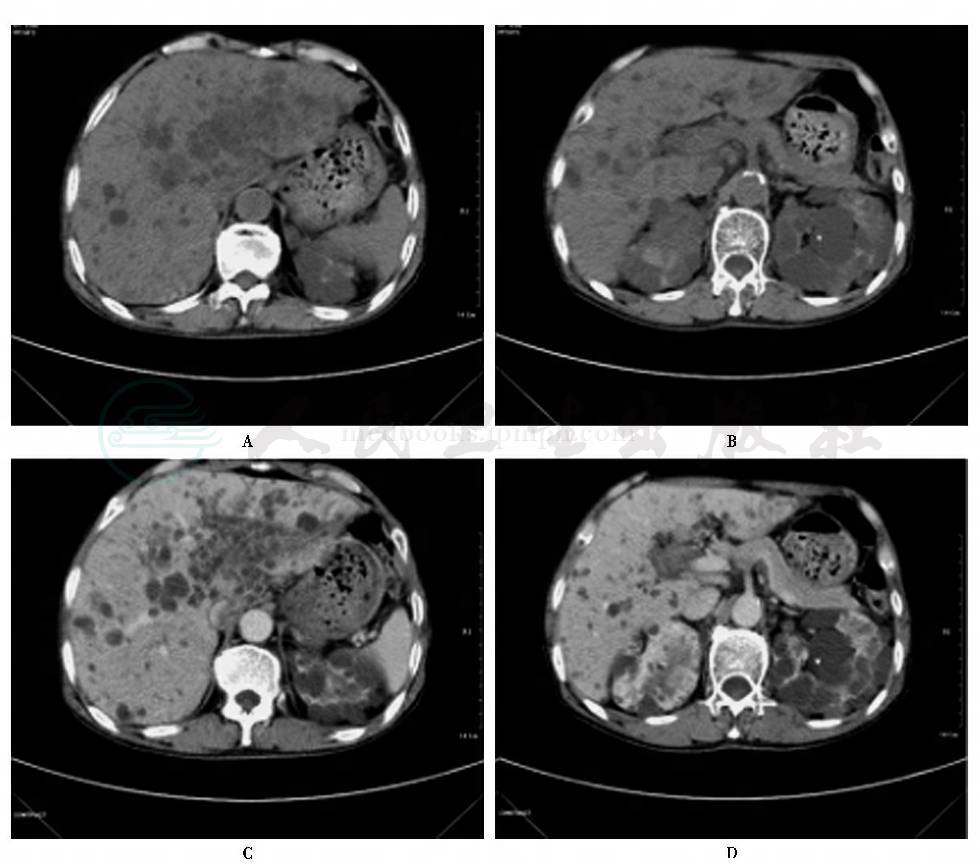
图4 ADPKP相关多囊肝CT平扫及增强
2.CT扫描
肝脏CT扫描均常规使用螺旋扫描技术,近年来螺旋CT扫描器的最大技术进展是多层扫描,多探测器扫描技术的优点是更快速及能获得很薄层的图像,典型的如现时肝脏扫描中所用的1.25mm、2.5mm、5mm厚图像。显示更薄的切面能提高对肝脏局灶性病变的发现率和诊断的准确性。在一次屏气时间内,可以探查更大的范围因而移动所产生的伪影可以最小,并且较薄的层面能减少容积平均伪影(volume averaging artifacts),因而可避免病灶被隐蔽和诊断错误。
CT扫描能准确显示肝囊肿的部位、大小、范围及性质,准确率达98%。CT平扫肝囊肿呈单发或多发圆形或卵圆形均匀低密度,边缘光滑锐利,CT值在0~20,囊壁薄而不能显示,除非有胆囊壁或肝包膜衬托,囊肿较小或合并出血和感染时,病灶CT值可增高。增强扫描后病灶无强化(图4)。
若是一个小的囊肿仅占据扫描层厚的一部分,根据部分容积平均数,其密度值应为囊肿与邻近组织的平均值,可误认为高密度病变。往往与实质性占位混淆,鉴别的方法为病灶层面薄层(2~5mm)扫描,最好作增强。如若囊肿的CT表现十分典型,除非临床病史提示囊性肿瘤或脓肿的可能,一般无需进一步检查。可能与良性囊肿混淆而需鉴别的病变包括一大类囊性病变,如罕见的原发性胆系囊性腺瘤,以及转移性平滑肌肉瘤、结肠癌、黑色素瘤、类癌,还有包囊虫病、陈旧性肝内血肿以及继发于胰腺炎的肝内假性囊肿等。囊壁局限性或广泛性明显增厚,壁内结节、分隔、密度不均匀的液体内容物以及造影后囊壁增强等特征都应引起人们注意,提示病变可能为非单纯良性囊肿。为明确诊断,经皮抽吸活检是需要的,超声检查也是有益的。
3.MRI
表现为T1WI为低信号,T2WI为非常明亮的高信号,如有出血,则T1WI和T2WI均为高信号。增强扫描无强化(图5)。
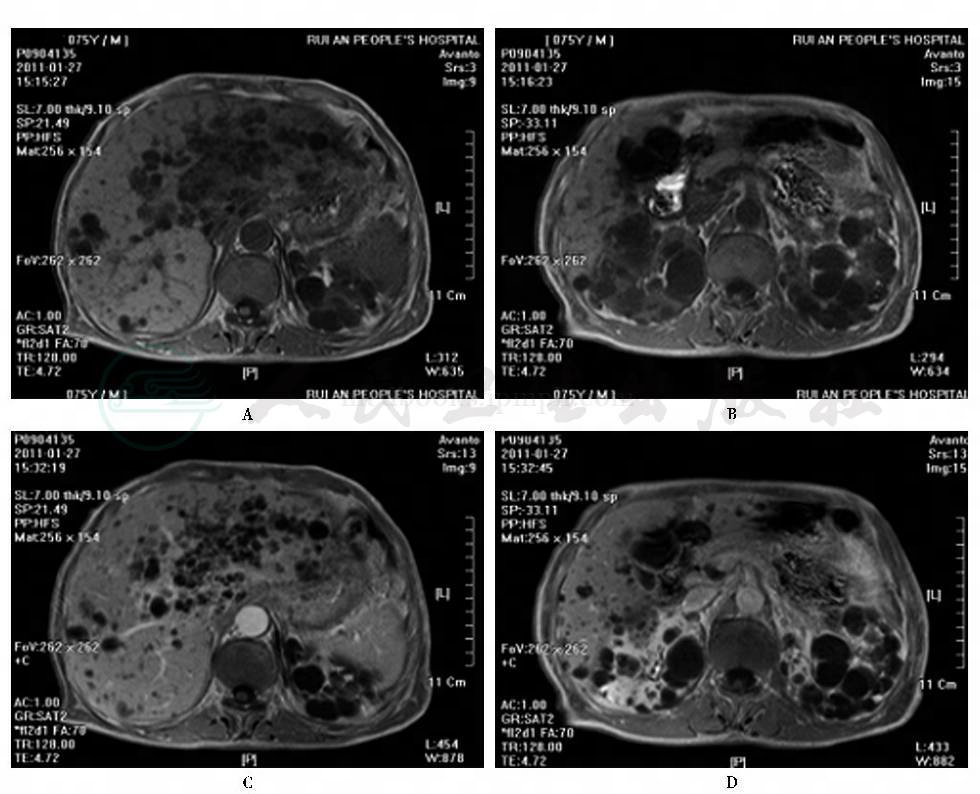
图5 ADPKP相关多囊肝MRI平扫及增强
4.分子诊断
ADPKD的分子诊断有很大进步,可直接检测基因序列。PKD1突变家族中PKD1突变检出为44%~76%,PKD2突变检出为75%。分子基因诊断是较新测试方法,尚缺统一的指南。基因检测对有ADPKD危险,年龄<30岁,无症状者而言有益。在此情况下超声诊断敏感性差,基因连锁分析,确认有无PKD1或PKD2突变,影响家庭计划或选择进一步细察。发现ADPKD突变也鼓励规律地血压监测和筛选联系的疾病如脑动脉瘤、二尖瓣脱垂。基因检测评估年轻者供体为ADPKD肾衰竭患者做肾移植,明确无突变,将进一步减少供体和受体的风险。欧洲已开展PCLD基因检测,包括PRKCSH序列检测,不久将有检测SEC63突变。
超声波检查肝和肾囊肿是明确囊肿表型的首选步骤,超声波诊断PCLD敏感性尚未肯定。PRKCSH检测鉴定无症状PCLD患者比ADPKD患者更有意义。基因检测结果不能立即改变PCLD患者处理。筛查家族成员潜在基因突变危险因素的无症状者是重要的,因患者常涉及子女危险,甚至不明突变状态,亦推荐正式基因咨询。
小、无症状肝囊肿无需处理,大而有压迫症状者应予以外科手术治疗。治疗方法包括囊肿穿刺抽液术、囊肿开窗术、囊肿引流术、囊肿切除术和肝切除术等。
1.囊肿穿刺抽液术
适用于浅表的肝囊肿、患者不能耐受手术的巨大肝囊肿(直径15cm以下)及合并感染的肝囊肿。在B超定位引导下经皮穿刺进入囊腔,尽量将囊液吸尽。大的囊肿需反复抽液,须注意避免继发感染。
2.囊肿开窗术
适用于直径15cm以上单发性肝囊肿、单发多房性肝囊肿、引起症状的多发性肝囊肿之中的大囊肿。手术在剖腹下或电视腹腔镜引导下将囊壁部分切除,吸净囊液,切缘仔细止血后囊肿开放。
3.囊肿引流术
适用于囊壁厚的肝囊肿。可行囊肿空肠Roux-Y吻合术,应注意预防肠内容物反流至囊腔引起继发感染。
4.囊肿切除术
适用于带蒂肝囊肿。
5.肝切除术
适用于并发感染后囊内出血或囊液混有胆汁且病变局限于肝脏的一叶者,巨大囊肿或多发性囊肿局限于肝段或肝脏一叶者。









