


 收藏
收藏 已收藏
已收藏先天性肛门直肠畸形占小儿先天性消化道畸形的首位,发病率在1/1500~5000,类型众多,直肠盲端和瘘管的位置各异。男性多于女性,高位畸形在男性约占50%,女性占20%。各种瘘管的发生率在女性为90%,男性为70%。合并其他先天性畸形的发生率约有30%~50%,且常为多发性畸形。仅1%有家族史,但遗传方式尚无定论。
肛门和直肠的发育,发生在第4周~6个月期间,胚胎长度为4~200mm阶段。在胚胎4mm时,后肠扩大与尿囊相通,形成泄殖腔,为一盲囊,中肾管开口于泄殖腔内。胚胎5mm时,泄殖腔与尾肠延伸相通。在体壁的腹侧有泄殖腔膜,系外胚层与内胚层相融合的很薄组织,使泄殖腔和体外相隔。第4周泄殖腔开始分成两部分,背侧部形成直肠,腹侧部称为尿生殖窦。其分隔过程是中胚层组织向尾侧方向生长,称为Tourneaux褶。间质从侧壁向内侧方向增生,称为Rothke褶。两种组织结构在中间融合形成尿直肠隔。
随着泄殖腔的分隔,泄殖腔膜也被分为前后两部分,前面为尿生殖窦膜,后侧成为直肠膜,并构成原始会阴。在第7周末,尿生殖窦向外开口,第8周时直肠肛膜破裂。在此之前,从第5周开始,外胚层向内发展形成肛凹,并逐渐加深接近直肠,最后两者相通。
直肠肛门畸形的发生是胚胎发育期发生障碍的结果,男性和女性基本上是相同的,仅是解剖上的区别。泄殖腔分隔过程的结果,尿生殖窦与肛门直肠窦之间相通,构成高位或中间位畸形,发生各种肛门直肠发育不全及直肠与尿道或阴道间的瘘管。肛门后移过程障碍和会阴发育不全的结果,构成低位畸形,发生肛门皮肤瘘,肛门前庭瘘,肛门狭窄等。
肛门直肠的局部解剖和排便控制机制是个复杂问题。新生儿肛管长度1.2cm(0.8~2cm),直肠长度5.2cm(3.5~7.5cm),近肛门处存在前凸的直肠会阴曲,肛管纵轴与会阴平面交角85°(60°~90°)。腹膜返折距肛门约2.9cm(2~4cm),骨盆神经丛位于骶前距肛门约3cm。直肠末端的环肌束向下延伸并增厚形成的括约肌,是平滑肌,呈不自主的收缩状态,以闭合肛门。环绕直肠的外括约肌是横纹肌,可随意志而收缩,包括尖顶袢、中间袢和基底袢三个肌群。Shafik认为肛外括约肌与耻骨直肠肌应视为一个统一体,共同构成控制排便的三个重要肌环,称为三环系统(triple loop system)。具有直接反向压迫作用和交锁机制,对肛门进行有力的控制。三环袢完全损伤,即导致肛门失禁。在肛门直肠畸形时,各肌纤维发育不正常,纤维走向亦有改变。Pena将外括约肌包括耻骨直肠肌肌纤维称为横纹肌复合体(striated muscular complex),以利手术进行。直肠耻骨肌在直肠肛管交接处形成一个环状吊带,称为耻骨直肠环(Puborectal sling),收缩时增加直肠下段的压力,起到排便的控制作用。盆腔肌左右两束肌群总称肛提肌,形成小骨盆腔内协调的收缩作用,帮助排便。联合纵肌是内、外括约肌间的纤维性组织,起固定肛管作用并协助排便。肛门直肠畸形手术时,应尽量保持肛门直肠周围肌群的完整性,以获得较完善的排便功能。
肛门直肠畸形的分类方法很多。1970年经Stephens倡议而制定的高位、中间位和低位的国际分类法,以直肠盲端与肛提肌和耻骨直肠肌的关系作为区分,在骨盆的侧位X线片上,从耻骨体中点至骶骨尾骨之间的连线,即耻尾线(PC线)作为耻骨直肠肌位置的标志(图1)。
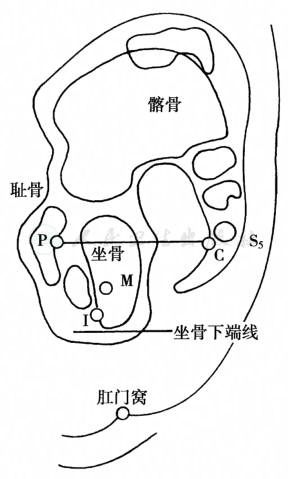
图1 X线诊断时的标准线
直肠盲端位于此线以上者为高位畸形,位于此线或稍下方为中间位畸形,低于此线者为低位畸形,同时又按性别、瘘管的有无、部位而分为许多亚型。由于此分类法过于复杂,影响临床应用,1984年世界小儿外科医师会议制定了比较简化的方案(Winspread直肠肛门畸形分类法),曾被大多数临床医师采用。
2005年在德国Krickenbeck举行了肛门直肠畸形治疗标准的国际会议,在回顾病因学、诊断和处理基础上,制定了新的规范的ARM分类,与Wingspread分类相比,该分类更便于不同手术方法之间的相互比较,目前国际上大多采用Krickenbeck分类法(表1)。
表1 ARM国际分类的诊断标准(Krickenbeck,2005)

先天性肛门直肠畸形常伴发其他畸形,发生率为28%~72%。目前一致认为高位肛门直肠畸形伴发畸形的发生率明显多于低位畸形,而且更加严重。伴发畸形最多的为泌尿生殖畸形,其次为脊柱,消化道、心脏等。有学者将肛门直肠畸形伴发畸形归纳为VACTER综合征(V:脊柱;A:肛门;C:心脏;T:气管;E:食管;R:肾脏和四肢)。
(一)泌尿生殖畸形
伴发泌尿道畸形最多见,上尿路畸形包括单侧肾脏缺如、肾发育不良、孤立游走肾、融合异位肾、马蹄肾、单或双侧肾积水、巨输尿管、膀胱输尿管反流等;下尿路畸形包括神经膀胱、膀胱外翻、尿道狭窄、尿道下裂等。男孩为女孩的2倍,高位畸形伴发的泌尿系畸形占60%,低位占20%。因此近年来提倡对肛门直肠畸形,尤其是中、高位的畸形患儿进行常规的泌尿系统检查,包括B超检查及排泄性膀胱尿道造影等,对有膀胱输尿管反流者需进行积极的抗感染治疗及密切随访。
(二)心脏畸形
由于发生肛门直肠畸形之时,恰在心血管系统发育的时期,因此伴发畸形亦多,占7%~12%,较一般小儿的发生率高20倍,最常见的是法洛四联症和室间隔缺损。
(三)胃肠道畸形
无肛伴食管闭锁是最常见的,发生率在10%左右。其他肠道畸形约有4%,如肠旋转不良和肠闭锁,因此,当发现消化道下段有畸形时,应仔细检查消化道上段。
(四)脊柱、四肢畸形
伴发脊髓和四肢畸形13.1%。脊髓的异常可由中央神经管及所在的脊髓、软组织的异常发育所致。在病变部位,脊髓可以发生粘连(拴系),从而阻止了它随锥体生长而上升,影响其末端部分的血供,导致肠道及膀胱的神经传递发生问题,有背部疼痛感及步态不稳等症状。脊髓拴系及各种形式的脊髓发育不良在3个月前的婴儿可通过无损伤的脊髓B超或MRI检查做出诊断。
(一)倒立侧位X线片
称为Wangensteen-Rice法,要求在生后12小时以上摄片,等待气体到达直肠,有时需要更长时间。在会阴肛门区皮肤上涂钡剂作为标记,摄片前将婴儿倒立2~3分钟,使直肠盲端的胎便与肠管气体互相转换,采取髋关节呈90°屈曲位,使保持能充分显示P点(耻骨中心)、C点(骶尾关节)、I点(坐骨最低点)的角度,以股骨大粗隆为中心摄片。
通过I点设一与PC线相平行的I线,与PC线间的距离为肛提肌群,直肠盲端位于PC线上方者为高位,于二线之间为中间位,超越I线为低位。或者设置M点,即坐骨结节的上2/3与下1/3交接点,在M线上方者为中间位,M线下方者为低位。
(二)瘘管造影
瘘管造影要求显示造影剂注入时的结肠影像及造影剂排出时的直肠瘘管影像。结肠直肠与尿道双重造影可显示直肠瘘管与尿道的关系。阴道造影可显示阴道与直肠的关系。
(三)超声波检查
患儿取膀胱截石位,探头接触肛门皮肤,在膀胱和骶骨锥体之间可见管状结构为直肠,内部呈无回声或稀薄胎粪的低回声以及含气体时可见强回声光团。测量管腔盲端至肛门皮肤的最近距离,>2cm为高位,1.5~2cm为中间位,<1.5cm为低位。
(四)磁共振成像
磁共振成像可立体分析肛门部肌群的形态、直肠肛门角。对于术前病例,MRI可提供高位、中位和低位等各型间肌群形态的比较,有利于手术时把握各个病例的特征;对术后病例,MRI可判断手术成功与否,如已成形直肠是否通过耻骨直肠肌,是否有效利用外括约肌等;对判断有无再次手术的必要,MRI起决定性作用。
先天性直肠肛门畸形的治疗主要是外科治疗,目的是重建具有正常控制功能的排便肛门,根据其类型和畸形的高低决定不同的手术方法。其治疗原则是为了改善术后排便控制功能,拖出的直肠必须通过耻骨直肠肌环,为了更好地识别耻骨直肠肌和尿道,中间位和高位畸形可采用经骶后矢状入路肛门直肠成形术(Pena)或经骶腹会阴肛门成形术。对中间位和高位畸形者,生后仍推荐先行暂时性结肠造瘘术,待至3~6个月时施行肛门成形术,术后3个月关闭造瘘。
经骶矢状入路肛门直肠成形术在直视下处理瘘管,以电刺激识别肌群的位置,保存直肠及肛周的肌肉神经血管组织,并恢复原状,如若直肠太短或太宽,则从腹腔游离及作尾状修剪,使直肠盲端准确通过肛提肌及括约肌群中央,从而得到满意的排便功能。目前亦有在腹腔镜下经腹会阴肛门成形术,其疗效有待进一步随访。
至于低位畸形,如肛门皮肤瘘无狭窄,排便功能无障碍者,不需治疗。肛门或直肠下端轻度狭窄,一般采用扩张术多能恢复正常功能。对肛门皮肤瘘者,仅作简单的“后切”手术。膜性肛门在新生儿期施行会阴肛门成形术。肛门前庭瘘如瘘孔较大,在一段时间内尚能维持正常排便,可于出生6个月以后施行手术。低位者因已通过耻骨直肠肌环,故手术较为容易,且术后排便功能良好。
至于泄殖腔畸形的治疗,因一穴肛畸形复杂,新生儿期先作暂时性结肠造瘘,6个月~1岁时行根治术。做成皮管或带蒂小肠移植的阴道成形术,在新阴道后方行腹会阴肛门成形术,利用原泄殖腔构成尿道一部分,进行泌尿系器官成形术,争取一期完成。









