


 收藏
收藏 已收藏
已收藏中文别名 :VHL病
VHL病(Von Hippel-Lindau disease)是一种常染色体显性遗传性多系统肿瘤综合征。临床表现以视网膜和中枢神经系统血管母细胞瘤、肾透明细胞癌、嗜铬细胞瘤、胰腺肿瘤或囊肿、附睾囊腺瘤等为主要特征,发病率约为1/36 000。VHL基因突变是其遗传学的发病基础,其中约20%患者为自身新发突变(de novo)。在此,我们仅描述VHL病患者中伴发嗜铬细胞瘤的部分。VHL病伴发嗜铬细胞瘤见图1、图2,其他常见肿瘤见图3。
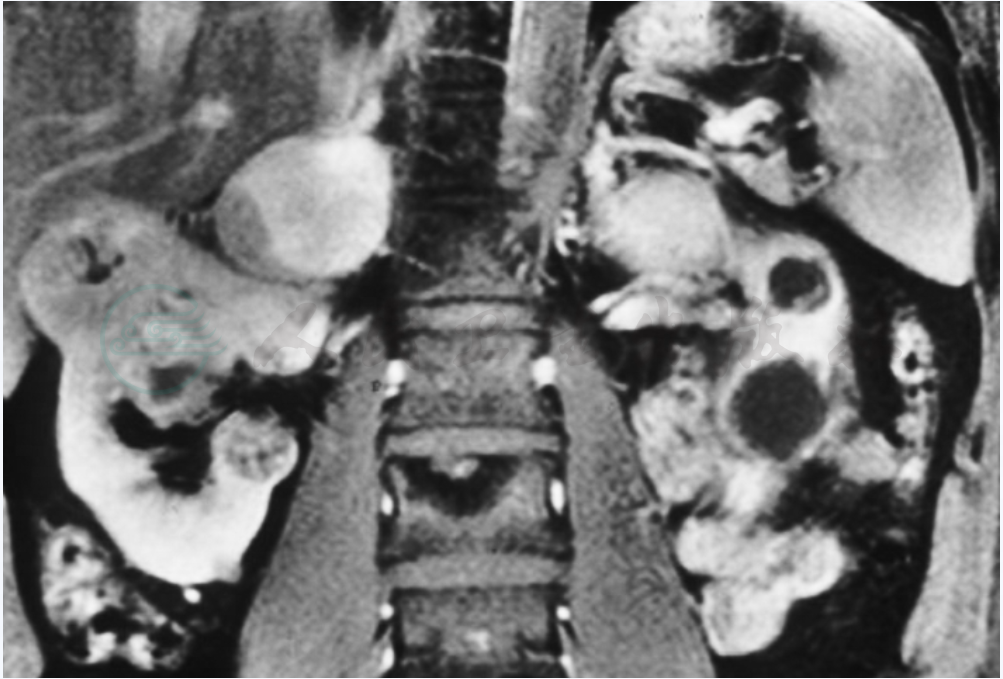
图1 患者34岁,MRI,VHL病伴双侧肾上腺嗜铬细胞瘤及双侧部分囊性肾癌
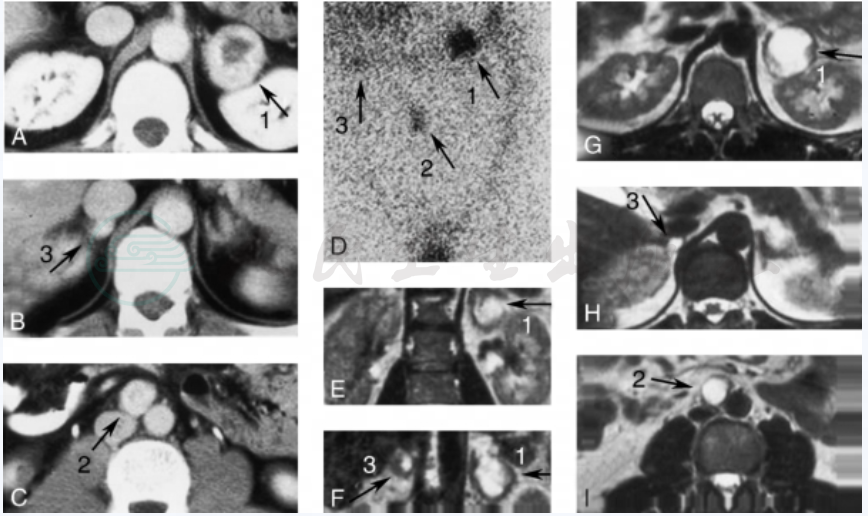
图2 30岁VHL病患者,伴发双侧肾上腺嗜铬细胞瘤(1、3)和腹部肾上腺外嗜铬细胞瘤(2)
A~C:CT;D:MIBG 显像(前视图);E、F:冠状位 MRI(前视图);G~I:水平位MRI。3个肿瘤全部腹腔镜切除
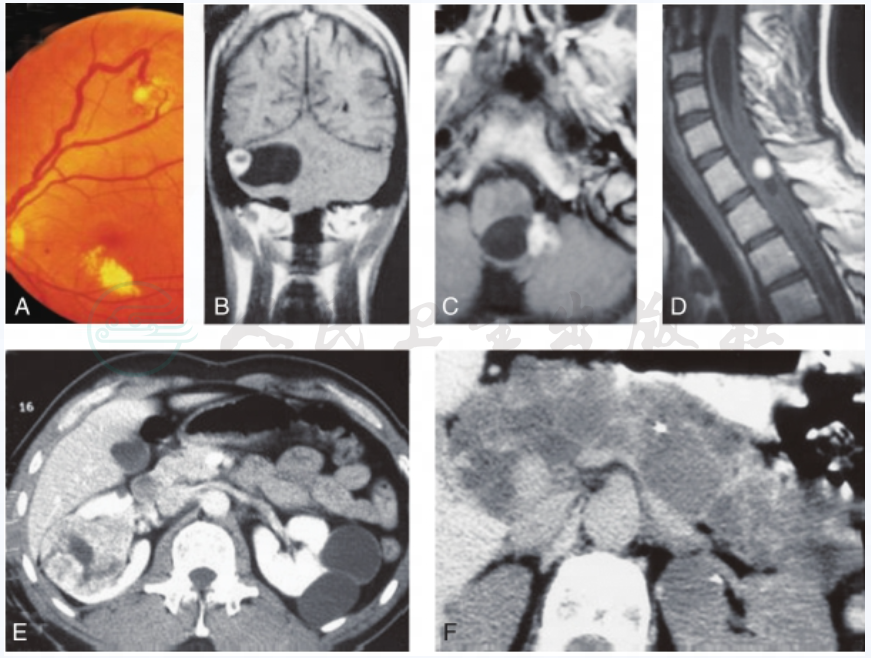
图3 VHL病中副神经节系统外的改变
视网膜血管瘤(A),中枢神经系统血管母细胞瘤:小脑(B,前视图)、脑干(C,俯视图)、脊髓、颈部(D,侧视图),肾癌、肾囊肿(E)和多发胰腺囊肿(F)
VHL基因突变相关嗜铬细胞瘤的发生率为10%~20%,通常位于肾上腺内,50%为双侧发病,恶性少见(<5%)。平均诊断年龄20~40岁,50%以上患者可同时伴发其他相关肿瘤。
目前已发现的多种VHL基因突变均可导致嗜铬细胞瘤的发生,涉及VHL基因所有的外显子。由于过低表达苯基乙醇胺-N-甲基转移酶,使绝大多数VHL病相关的嗜铬细胞瘤仅分泌去甲肾上腺素。通常患者血清尿中仅见去甲肾上腺素的代谢产物3-甲氧基去甲肾上腺素单独升高。
根据是否伴发嗜铬细胞瘤,VHL病分为两个不同类型:Ⅰ型(具有除嗜铬细胞瘤以外的上述其他肿瘤)和Ⅱ型(常伴发嗜铬细胞瘤)。Ⅱ型又可分为:大多不伴有肾癌(ⅡA型),常伴发肾癌(ⅡB型)及几乎只伴发嗜铬细胞瘤(ⅡC型)三种亚型。其中Ⅰ型中的VHL基因以大片段缺失、截断突变为主;而与嗜铬细胞瘤相关的Ⅱ型则以错义突变为主。其热点突变为VHL基因第3外显子的第167位p.R167W和p.R167Q突变,其中p.R167W突变与ⅡB型密切相关。
预防医学在VHL病的防治中发挥重要作用,VHL病中大部分肿瘤包括视网膜血管瘤(激光疗法),小脑、脑干和脊髓的血管母细胞瘤(神经外科切除),肾癌(保留器官手术)和嗜铬细胞瘤(腔镜手术)等通过早期的诊治可获得较好的疗效。









