


 收藏
收藏 已收藏
已收藏英文名称 :incontinentia pigmenti
内容提要
● 本病由Xq28区NEMO基因(编码NF-иB必需调节子)突变所致,男婴通常致死。
● 皮损沿Blaschko线分布,可分为四期,即炎症期、水疱期、疣状期、色素沉着及色素减退期/萎缩期。
● 其他外胚层异常可能存在,如脱发、指甲营养不良、楔形牙等,偶尔累及眼睛,骨骼和神经系统。
色素失禁症(incontinentia pigmenti,IP)是一种少见的X连锁显性遗传的皮肤色素异常疾病,男性常在子宫内死亡,主要累及女性,表现为新生儿散在分布的炎症性水疱性损害,数月后变成疣状损害,最后则变为色素沉着。约80%患者可发生其他部位的先天性畸形,如有眼、中枢神经系统、牙及骨骼系统的损害。Garrod于1906年首先报道本病。
1.基因突变
已证实有两个色素失禁症位点,位于X染色体长臂的 Xq11(IP1)和 Xq28(IP2)。核因子(NF)-кB 基因调节体(NEMO)基因突变在抑制肿瘤坏死因子(TNF)诱导的细胞凋亡中起作用,显示是本病发生的原因。该病被分为IP1(散发型)和IP2(遗传型)。国际色素失禁症联盟确认IP2是由于NEMO基因突变(NF κ B基本调节器)引起(图27-23)。这种基因的突变在男性患者常为致死性,但若男性患者患有Klinefelter综合征则有可能存活。在部分男性病例中嵌合体也可能起作用。
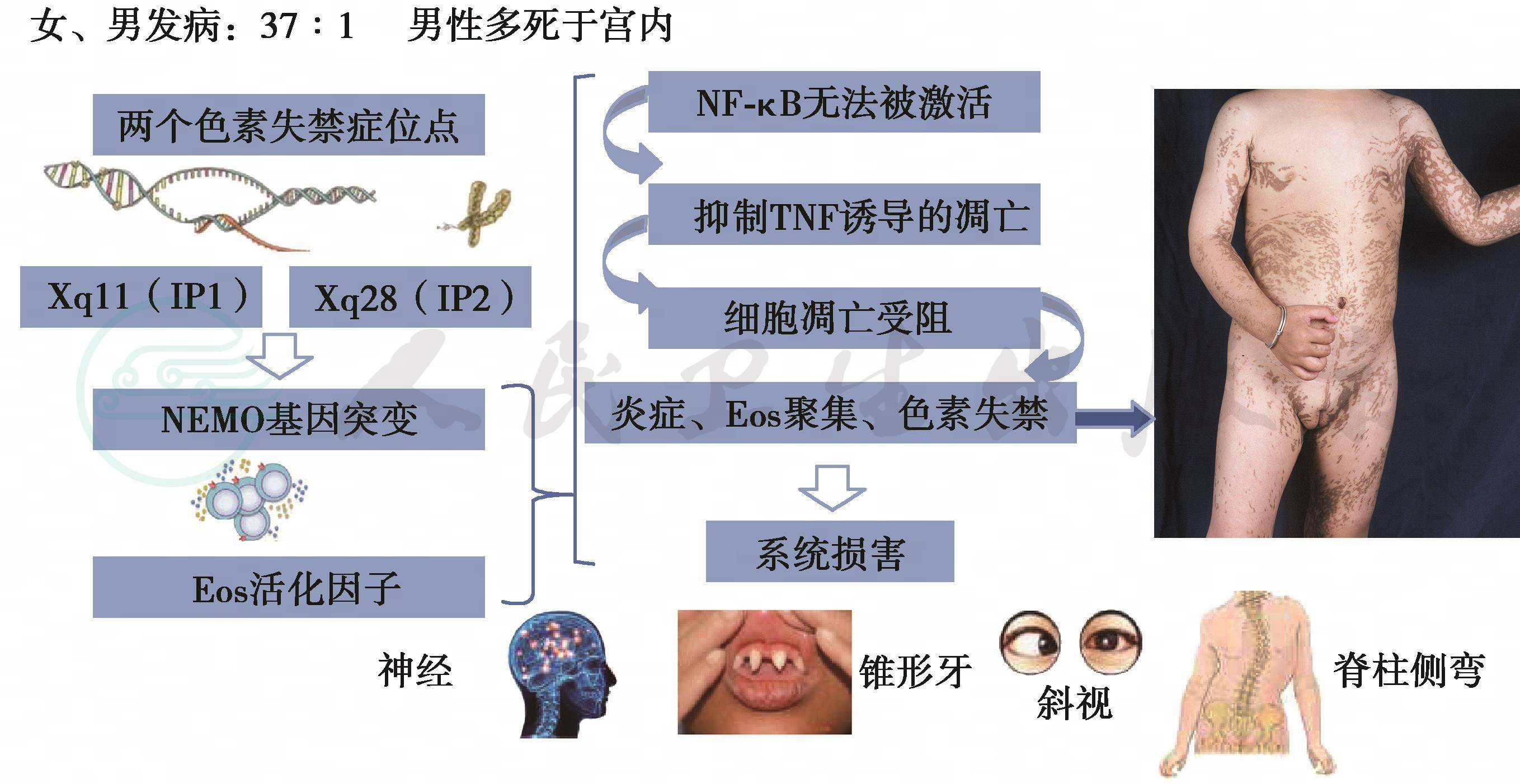
图1 色素失禁症发病机制
注:Eos=嗜酸性粒细胞;NF κ B=活化的趋化因子;NEMO=NF-κ B 基因调节因子;色素失禁有两个基因突变位点,由染色体Xq28 的NEMO 突变引起,EOS 活化因子释放,由于缺乏NEMO,造成NE κ B 无法被激活,细胞凋亡(受阻)产生炎症、EOS 聚集、色素失禁病变。第Ⅲ期,凋亡增加,由正常皮肤取代。
2.其他因素
嗜酸性粒细胞趋化因子、角质形成细胞源性白三烯B4、免疫反应的变化皆为本病发病的因素。
1976年Carney统计分析了世界有关文献的653例患者,发现55.4%的患者有家族史。男女比例是1∶37,因而推论本病属于X连锁显性遗传。患有色素失禁症的妊娠妇女有25%的自发性流产(男性受累)风险,大多数男性患儿死于宫内,仅有少数存活。其女性患儿50%将受到影响。比其母亲受累更为严重。
各期皮肤组织病理变化:第Ⅰ期表皮内海绵形成水疱,含有多量嗜酸性粒细胞。第Ⅱ期疣状皮损不规则乳头瘤样增生;角化不良细胞排列成旋涡状。第Ⅲ期真皮上部嗜黑素细胞内黑素沉积,伴有基层色素减退。第Ⅳ期表现为表皮变薄和真皮附属器缺如。
本病皮肤损害可以自愈,一般无需治疗。医师的职责是监测其疾病过程,治疗仅控制水疱损害的继发感染。
本病皮肤病变有逐渐减少之趋势,多数病例的皮肤改变可以恢复,色素自然消退。如伴有中枢神经系统损害或眼的病变,以及骨、齿、发的改变,常不随皮肤好转而好转,通常采用对症治疗。
用0.1%依沙吖啶液外搽或局部使用2%莫匹罗星软膏。皮损严重,可口服中小剂量糖皮质激素。
系统损害 防治癫痫发生,白内障复明、唇腭裂修补、牙列不良正畸。









