


 收藏
收藏 已收藏
已收藏英文名称 :Langerhans cell histocytosis
中文别名 :组织细胞增生症-X;Letterer-Siwe病;Hand-Schuller-Christian病;孤立性嗜酸性粒细胞肉芽肿
朗格汉斯细胞组织细胞增生症(langerhans cell histiocytosis,LCH)又称为组织细胞增生症 X(Histiocytosis X);组织细胞增生症Ⅰ类,是由朗格汉斯细胞克隆性增生形成的一组反应性疾病,可以发生在全身各个器官、系统或组织而引起的相应的损伤。该组疾病可以相互重叠,并且由于其临床经过的严重程度不等而形成谱系,即可以表现为温和的、无症状的单器官受累,也可表现为严重的、进行性的多器官受累性疾病,但是该组疾病共同特点是增生的细胞表达S-100和CD1a蛋白,胞浆内可见Birbeck颗粒。1987年,组织细胞协会将该组疾病统称为朗格汉斯细胞组织细胞增生症。
LCH病因尚不完全清楚。关于LCH为炎症性疾病还是真正的肿瘤性疾病,仍存有争议。LCH病变中除了不同比例的克隆性树突状细胞外,还存在巨噬细胞、淋巴细胞、嗜酸性粒细胞、多核巨细胞、中性粒细胞及浆细胞等多种炎症细胞的浸润。病理性LC和皮肤正常LC虽然有相似之处,但功能却有差别,表达异常细胞黏附分子,而抗原呈递功能减弱。直到2010年,研究报道了57%的LCH病变中发现了BRAF V600E突变,高度支持LCH是一种克隆性肿瘤性疾病。近些年来也有研究认为LCH是一种非成熟LC单克隆性疾病,来源于骨髓造血干细胞及髓样前体细胞,且这些细胞中检测到BRAF V600E或其他RAS / RAF / MEK(MAPK / ERK信号通路途径)路径相关基因突变。故目前研究认为LCH是以CD1a+ CD207+树突状细胞浸润的髓源性肿瘤疾病。
LCH是一个罕见疾病,据统计本病在全世界的发病率约4/1000000~5 4/1000000。由于LCH可以发生在不同的组织/脏器和皮肤,因此,实际数据可能高于该统计数据。本病可以发生于任何年龄,但1到3岁儿童多见。在美国,每年儿童发病率约 0 5/100000。男女比例是 2∶1。
研究者关于LCH的发病机制有多种观点。
1.克隆增殖异常
有研究通过X染色体连锁的DNA探针证实病理性朗格汉斯细胞(pathologic Langerhans cells,pLCs)均为单克隆性CD1a阳性的组织细胞,而病变中的T细胞则为多克隆性,提示本病为朗格汉斯细胞单克隆性增生所致克隆增殖性疾病,其生物学行为多样,有肿瘤的部分特征。但LCH的生物学行为和肿瘤又不完全相同。另外,近年的研究发现该病可能与抗原提呈细胞的祖细胞发生癌基因突变有关,在LCH患者的病变组织中发现了BRAF V600E突变蛋白。而且,在接受BRAF基因突变药物达拉非尼/ 维莫非尼治疗后,临床情况迅速好转,提示了LCH患儿存在克隆增殖异常。
2.细胞因子介导
最近有人通过对LCH的病理参数、Ki-67增殖和核分裂的评估,提出LCH也许是反应性病变,可能是某些细胞因子引起的免疫反应。LCH患者的 TNF-α、IL-1、IL-6、粒-巨噬细胞集落刺激因子及白血病抑制因子显著增高,Dina等曾用单克隆抗体对一些LCH患者的血管内皮生长因子(vascular endothelial growth factor,VEGF)测定发现,70%的LCH患者表达VEGF,在多系统受累的播散型LCH中其水平明显升高,故有学者提出LCH的发病机制还和一些细胞因子的介导有关。
3.染色体异常
有研究发现LCH细胞染色体有异常的改变,包括染色体和染色单体的断裂,且在多器官受累的患者中此种染色体异常的数量比单器官受累的患者显著增多,Scappaticci等认为这种染色体的不稳定性可能是由于本身遗传的不稳定性造成的,也可能是对外界因素刺激(如病毒)的反馈,它可能在LCH的发病机制中起重要作用,甚至成为恶变的危险因素。还有一些E-钙黏蛋白等维持内皮细胞完整性的细胞黏附分子的突变,阻碍了朗格汉斯细胞向淋巴结的正常迁移,而导致LC异常增殖聚集,故认为E-钙黏蛋白和LCH发病机制有关。
4.其他
LCH发生可能还与病毒感染(如人疱疹病毒6型和EB病毒)、接触石棉及免疫调节功能紊乱等因素有关,成人肺LCH几乎均与吸烟有联系,此外,也有学者认为LCH与遗传有关,但关于本病家族性聚集的报道很少。
临床上,LCH多表现为三种形式,相互之间有部分重叠。①孤立性嗜酸性肉芽肿:大多数病例为这一型,表现为孤立性病灶,通常累及骨(颅骨、股骨、盆骨或肋骨等)、淋巴结或皮肤;②Hand-Schuller-Christian病:系单系统、多灶性疾病,大多累犯骨骼系统;③Letterer-Siwe病:疾病累及多器官、多部位,常见受累部位包括骨、皮肤、肝、脾和淋巴结。
病灶一般较小,数毫米至数厘米之间,病灶一般位于髓腔内,呈红色或橙色软组织,软组织可突破骨皮质。镜下朗格汉斯细胞呈中等大小,胞浆边界不清楚,嗜酸性,细胞核椭圆形,边界不规则,有时呈锯齿状,常见核沟。染色质可分散于细胞核内或聚集于核膜边缘。朗格汉斯细胞多排列成巢状,与炎症细胞混合,包括大量嗜酸性粒细胞、淋巴细胞、中心粒细胞和浆细胞。病灶中可见坏死,可见多核的破骨细胞样巨细胞。细胞表现相对较为旺盛的有丝分裂,每10个高倍视野中可见5~6个核分裂象。朗格汉斯细胞具有特征性的免疫组化表现,细胞膜可表达CD1α,细胞核及胞浆表达S100。这些细胞通常不表达CD45。
1.实验室检查
与患儿受累器官及病情危重程度有关。骨髓受累可出现血常规变化,如贫血、血小板减少及粒细胞减少等。脾脏明显肿大者也可出现全血细胞减少。血浆免疫球蛋白除IgM以外大多正常,补体C3可以降低。肝脏受累可出现转氨酶增高、胆红素升高及白蛋白降低、凝血功能障碍等。肺部受累患儿可以出现氧分压降低、肺功能异常等。
2.病理学检查
是本病的确诊依据。可做皮疹、淋巴结及齿龈肿物的活组织检查或病灶局部穿刺物或刮出物的病理检查。LC细胞质呈现均匀粉色,核弯曲呈咖啡豆样,细胞直径约13µm。电镜下可见网球拍或棒状的伯贝克颗粒。组织化学染色S100阳性,并与花生凝集素和CD1a单克隆抗体发生反应。
3.其他检查
本病的表现多种多样,错综复杂,需对患儿进行全面检查,包括影像学表现(X线、CT、MRI和PET等),确诊的关键在于病理检查发现朗格汉斯细胞的组织浸润,从而作出正确地临床诊断和分级。
LCH部分病例不经治疗可自行缓解,但某些病例(尤其是婴幼儿多系统LCH)如不采用联合化疗则进展很快,预后不良。目前国际组织细胞协会强调综合考虑各种危险因素,结合临床分类及预后等,制定了新的诊疗方案。
单系统疾病,包括单一或多发的骨骼、皮肤或淋巴结等病变,部分可自愈,多数可治愈,预后良好。如局灶性骨骼病变可单纯病灶刮除,无需全身化疗。此外,对于单系统组有“中枢神经系统危险部位受累”“多个骨受累”“特殊部位”及多系统组受累的患儿则需要系统化疗。
1.一线化疗及疗效评估
泼尼松(PRED)+ 长春花碱(VBL)诱导方案:VBL 6mg/m2,静脉注射,每周 1次,共 6周,PRED 每天40mg/m2,分3次口服,持续4周后减停2周,治疗前6周后,评估病情变化,并相应地继续治疗。治疗反应的评估主要分为3种,分别为良好(疾病好转或者痊愈)、混合(疾病稳定或原有病灶好转,但出现新发病灶)及进展(疾病恶化)。
如果有危险器官受累诱导6周后无好转或无危险器官受累治疗后出现危险器官受累的患儿,需尽早进入补救治疗,其他患儿继续诱导治疗6周,剂量为VBL 6mg/m2,静脉注射,每周1次;PRED 40mg/m2,分3次口服,每周口服3天。诱导治疗12周后,再进行评估。如仍有危险器官受累的患儿进入补救治疗。无危险器官受累但患儿病情仍无好转,则进入二线治疗。如果诱导治疗6周或者12周评估病灶完全消退的患儿可进入维持治疗,总疗程为12个月,具体方案为:VBL 6mg/m2,每3周1次,PRED每3周口服5天,6-巯基嘌呤(6-MP)每天50mg/ m2,口服至疗程结束。
2.二线化疗
对于难治性血液系统受累或肝功能障碍患儿,或者诱导治疗反应不好的患儿,可选择二线化疗。包括VBL+PRED联合克拉屈滨(2-CdA)或阿糖胞苷(Ara-C)的方案。
3.补救治疗
主要用于诱导反应不好的有危险器官受累的患儿,可采用低强度预处理造血干细胞移植方案或2-CdA联合Ara-C治疗。
4.靶向治疗
已有研究表明,LCH由MAPK / ERK通路中基因突变导致该信号通路的激活,且LCH神经变性病患儿活检中发现BRAF V600E细胞弥漫在血管周围,这一现象为靶向治疗提供了强有力的理论依据。因此,针对性靶向抑制该信号通路的活化,可能是LCH治疗的关键。自2015—2019年不同研究报道了使用达拉菲尼(BRAF V600E抑制剂)、维莫非尼或曲美替尼治疗LCH后,病情得到控制,治疗有效率为60%~75%。但由于LCH病因尚不明确,靶向治疗是否会影响LCH发展仍需要长期的追踪及调查。
5.其他相关治疗
部分医生认为放射治疗对青少年单一骨损伤可能是有用的,但放疗存在后遗症的风险,如放疗会使局部发生恶性肿瘤的风险增加。此外,LCH的各种并发症的治疗也非常重要。如目前认为尿崩症的有效治疗方法仍然是1-脱氨基 -8D-精氨酸加压素(DDAVP)替代治疗。生长激素缺乏导致的生长发育迟滞可用生长激素替代治疗。
附:LCH诊治流程图
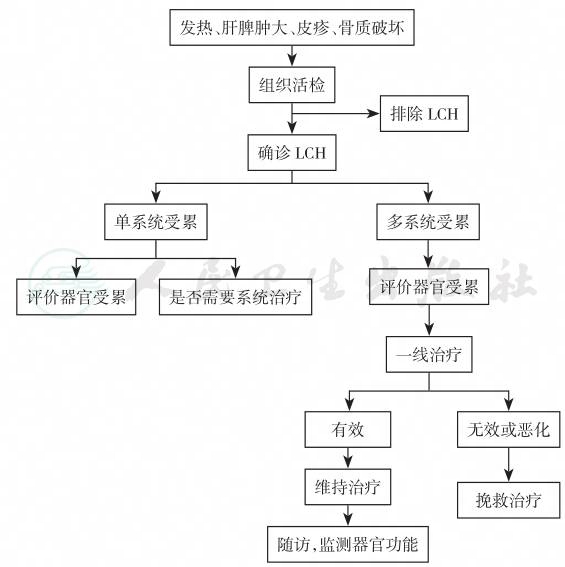
引自:儿童血液系统疾病诊疗规范(第2版).第1版.ISBN:978-7-117-33957-5.主编:
X染色体连锁的雄激素受体基因研究(HUMURA)已证实该病系朗格汉斯细胞的单克隆性增生。Ig和TCR基因均呈胚系构型。









