


 收藏
收藏 已收藏
已收藏英文名称 :infantile cortical hyperostosis
中文别名 :Caffey病
婴儿骨皮质增生症(infantile cortical hyperostosis,ICH),又称Caffey 病,是一种婴儿时期侵犯骨骼及及其邻近肌肉、筋膜的疾病,其特点为长管状骨和扁平骨有骨膜下新生骨形成,以及患处的肿胀和疼痛。由于本症少见,首诊时易误诊。
ICH 的病因至今未明,可能与骨骼感染、遗传、过敏因素有关。该症可多个部位同时受累,累及肌肉筋膜。主要发生于长管骨、下颌骨(70%~90%)、锁骨(50%)和肩胛骨、肋骨(20%~30%),除指/趾骨、脊柱及髋骨外全身骨均可累及,可单骨或多骨,多发者常表现为对称性特点,下颌骨多位于下颌角和下颌升支。病变骨和软组织内的小动脉内膜呈增生改变,造成缺氧可能是产生反应性骨膜增生的原因。亦有认为与先天性骨膜小动脉发育不良和过敏因素有关。近年报道认为ICH与COL1A1 基因突变有关。主要为编码Ⅰ型胶原蛋白α-1链(COL1A1)的基因错义突变杂合子(3040C→T),导致Ⅰ型胶原蛋白α-1链螺旋区Arg被Cys替换,易于触发骨皮质胶原纤维的急性炎症。
该症有两种类型:家族性ICH和胎儿期发生的致死性ICH。前者是一种暂时性的婴儿骨皮质增生的疾患,多见于5个月内婴儿,男孩多见,为自限性疾病,预后好。后者非常罕见,患儿多为死产或生后不久死亡。
ICH病理改变早期为骨膜水肿增厚,与邻近组织分界不清。亚急性期骨膜组织结构重新恢复,骨膜下新骨形成,骨皮质增厚。晚期增厚骨皮质由内至外逐渐吸收变薄,骨髓腔扩大,最后恢复正常。通常病变持续2~3周至2~3个月的骨皮质增厚,多在2岁之内消退,累及骨可遗留轻度的骨皮质增厚。
1.实验室检查
ICH病理上为急性炎性反应,临床常有白细胞增高,血沉增快,碱性磷酸酶增高等,可伴有贫血。
2.影像学检查
ICH的X线表现可分为软组织肿胀期、增生期和恢复期,X线表现特点主要体现在后两期。其特点为与骨病变范围相一致的软组织肿胀,与脂肪层分界清楚;各种形状诸如线状、带状、花边状等的骨膜增生,骨皮质多正常,增生的骨膜可形成“包壳征”或“管套征”。病变于长骨多见,且限于骨干。急性增生显著者典型X线表现为大范围的骨膜套管状增生、骨干呈梭形改变,骨膜下大量新生骨形成,病骨变粗。开始时骨皮质外新骨边缘毛糙,原有皮质的界限尚可见,随着新骨的密度增加则与原有的骨皮质融合(图1)。这种新骨形成围绕骨干呈对称性,不超过骨骺缘,反复加剧和消退。消退时原增厚的骨皮质可见骨质吸收、变薄但无破坏及硬化。随骨厚度的减少其临床症状也逐渐消失,数月或数年后病变完全吸收。各部位骨骼的病情可各异,累及不规则骨硬化较明显、骨膜呈环状增厚、包壳样骨化,但干骺端不累及可与其他骨膜增生性疾病鉴别。
CT表现除上述改变外可见明显软组织肿胀、边界不清。MR骨膜增厚及软组织水肿表现早于X线平片。M R特征主要表现为长骨以中段为主的明显骨膜套筒样增厚,信号特点为稍长T1、长T2,周围软组织呈长T1、长T2水肿信号,髓鞘也可见轻度水肿信号。高场MR对ICH有独到的诊断与鉴别诊断意义,能更早期的发现骨内外膜、骨间膜增生病变及其演化,并显示特征性“外星人眼征”和外层反应性纤维层等特征性影像改变,且可鉴别一些易于混淆的骨病变,以及早期识别和诊断ICH。
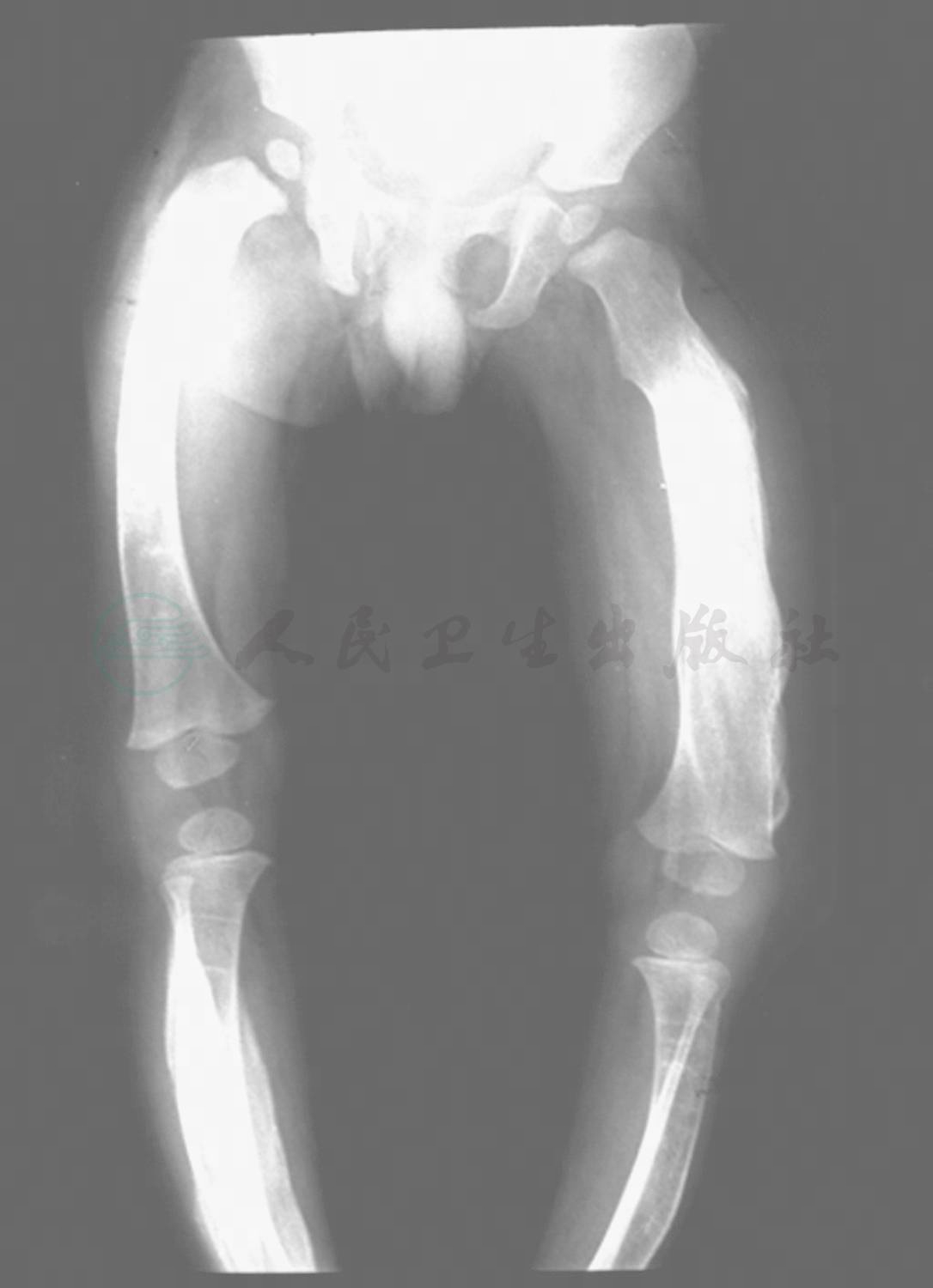
图1 婴儿骨皮质增生症双下肢正位片
右侧股骨、胫骨及左侧股骨骨膜明显增厚,外形清楚,无明显骨皮质破坏征,两侧干骺端形态正常,病变部骨质周围软组织肿胀、密度增高
ICH无需特殊治疗,一般预后良好。本症可以自愈,临床症状多在1个月内消失,实验室检查亦随之恢复正常。骨质一般6~9个月恢复正常,但也有少数反复发作,持续多年,慢性迁延后可遗留肢体畸形、运动障碍等。肾上腺皮质激素可缓解急性期的全身症状,适用于病变广泛的病例,但对骨修复无明显效果,且停药后易复发。









